
Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HCT) is an effective immunotherapy for human cancer. More than 20 000 allo-HCTs are performed each year worldwide, primarily for the treatment of hematologic malignancies. Several technical innovations implemented in allo-HCT over past 2 decades have reduced NRM by 50% and improved overall survival. The allo-HCT practice has changed with the introduction of peripheral blood, cord blood, and haploidentical transplantations and reduced-intensity conditioning, and the patient population is also different regarding age and diagnosis. However, both acute and chronic GVHD remain serious barriers to successful allo-HCT and it is not clear that a major improvement has occurred in our ability to prevent or treat GVHD. Nevertheless, there is an increasing knowledge of the biology and clinical manifestations and the field is getting better organized. These advances will almost certainly lead to major progress in the near future. As the long list of new potential targets and respective drugs are developed, systems need to be developed for rapid testing of them in clinical practice. The current reality is that no single agent has yet to be approved by the US Food and Drug Administration for GVHD prevention or therapy. Although a primary goal of these efforts is to develop better therapies for GVHD, the ultimate goal is to develop treatments that lead to effective prevention or preemption of life-threatening and disabling GVHD manifestations while harnessing the desirable graft-versus-tumor effects.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HCT) is an effective immunotherapy for human cancer.1 More than 20 000 allo-HCTs are performed each year worldwide, primarily for the treatment of hematologic malignancies. Several technical innovations implemented in the past 2 decades have reduced nonrelapse mortality (NRM) by 50% and improved the overall survival (OS) after allo-HCT.2 Observed decreases in mortality could be due to better methods for the prevention and treatment of GVHD, but to many other advances, including: better treatment of infection, less toxic conditioning regimens, and better HLA matching of unrelated donors (URDs). Allo-HCT clinical practice has also changed over last 20 years and has departed from the uniform use of HLA-matched sibling donor BM transplantations and myeloablative conditioning to a much more complex field. The introduction of peripheral blood, cord blood, and haploidentical transplantations and reduced-intensity conditioning (RIC) regimens, an older patient population, and different diagnoses have modified and made it more difficult to study factors that affect the risks and incidence of GVHD in today's era.3 Nevertheless, acute GVHD (aGVHD) and chronic GVHD (cGVHD) remain a major contributor to transplantation-related deaths and the most significant barrier to the success of allo-HCT.4–6 Despite prophylactic treatments with immunosuppressive agents, approximately 50% of transplantation recipients develop GVHD. Most GVH reactions are undesirable and affect multiple organs; however, GVH reactions against hematopoietic tissue targets are desirable and critical for the cure of hematologic malignancies (ie, the graft-versus-tumor effect [GVT]) and for donor immune-hematopoietic system engraftment. These disparate effects of GVH reactions are difficult to separate and any strategies directed against GVHD may adversely affect survival by increasing malignancy relapse or infections. This chapter examines the progress made in GVHD prevention and therapy. Other areas of progress, such as GVHD's impact on health-related quality of life and functional status and advances in basic research or trial designs, will also be discussed.
Who gets GVHD and how is it diagnosed?
GVHD is an immunological complication of allo-HCT caused by donor T cells recognizing the genetically disparate recipient who is unable to reject the donor graft.6 cGVHD is additionally complicated by disturbances in pathways of immunological reconstitution and failure to acquire immunological tolerance, thereby resulting in both alloimmune and autoimmune attacks on multiple host tissues.7
aGVHD diagnosis should be confirmed by biopsy of an affected organ if possible; in addition, other non-GVHD complications involving the skin, liver, and GI tract should be ruled out.8 Although diagnostic biopsies are highly specific if current histopathology criteria are used, the sensitivity of these biopsies is only approximately 60%; therefore, the ultimate aGVHD diagnosis and decision to treat systemically is based on careful integration of all available clinical information.9 There is clearly an unmet need for developing more accurate diagnostic tests for aGVHD.10 The severity of aGVHD is graded according to the Keystone 1994 consensus criteria (grades I-IV) or, less commonly, by the Center for International Blood and Marrow Transplant Research (CIBMTR) criteria (grades A-D).11,12 The diagnosis of cGVHD is also primarily clinical and requires at least one diagnostic sign in a target organ per National Institutes of Health (NIH) criteria (ie, a sign found only in cGVHD) or at least one distinctive sign (ie, a sign highly suggestive of cGVHD) in combination with some other laboratory, biopsy, or other test confirmation in the same or another organ.7 Due to the frequent presence of typical clinical manifestations, biopsies are less commonly done for cGVHD diagnosis and are more often used to rule out other diagnoses such as infection, drug reactions, or cancer.
The incidence of GVHD described in the available literature must be interpreted in light of new classifications that view GVHD as a continuum process rather than as a strict separation of aGVHD and cGVHD by the previously used day-100 posttransplantation cutoff. (Figure 1) The current consensus is that clinical manifestations rather than time after transplantation should determine whether the clinical GVHD syndrome is considered acute or chronic.7 Some signs and symptoms are common to both aGVHD and cGVHD (ie, erythema, macular-papular rash, nausea, vomiting or diarrhea, and elevated liver function tests) and thus cannot be used to distinguish the two. Two main categories of GVHD are now recognized, each with 2 subcategories. The broad category of aGVHD includes: (1) classic aGVHD (ie, macular-papular erythematous rash, gastrointestinal symptoms, or cholestatic hepatitis), occurring within 100 days after transplantation or donor leukocyte infusion and (2) persistent, recurrent, or late aGVHD, occurring beyond 100 days after transplantation or donor leukocyte infusion. To facilitate reporting in clinical trials, the arbitrary day-100 distinction is retained for the purpose of separating of these 2 aGVHD categories. Both aGVHD subentities occur without the presence of diagnostic or distinctive cGVHD manifestations. A second broad category is cGVHD, which encompasses: (1) classic cGVHD, which consists only of manifestations that can be ascribed to cGVHD; and (2) aGVHD and chronic overlap syndrome, in which features of both aGVHD and cGVHD appear together. With appropriate stratification, patients with persistent, recurrent, or late aGVHD or overlap syndrome can be included in clinical trials with patients who have cGVHD. The newly defined entities of “late-onset” aGVHD and overlap syndrome subset have been associated with poor survival in some studies but not in others.13–16 It remains to be determined whether the type or duration of immunosuppressive therapy should differ in patients with “classic” versus “late” aGVHD or “overlap cGVHD.”

GVHD classification after the NIH consensus conference. The current consensus is that clinical manifestations and not the time after transplantation determine whether the clinical syndrome is considered aGVHD or cGVHD. Retrospective and prospective studies reported wide ranges in the incidences of “late aGVHD” (3%-48%) and “cGVHD overlap” (13%-82%); more prospective cohort studies are needed.
GVHD classification after the NIH consensus conference. The current consensus is that clinical manifestations and not the time after transplantation determine whether the clinical syndrome is considered aGVHD or cGVHD. Retrospective and prospective studies reported wide ranges in the incidences of “late aGVHD” (3%-48%) and “cGVHD overlap” (13%-82%); more prospective cohort studies are needed.
Historically, cGVHD severity was staged as “limited” (ie, localized skin involvement and/or liver dysfunction) or “extensive” (ie, generalized skin involvement, liver histology showing aggressive hepatitis, or involvement of any other target organ).17 This classification is relatively poorly reproducible across investigators and does not provide information about the number and extent of the organs involved or the severity of organ function impairments.18 A new cGVHD clinical staging system is now recommended for scoring of individual organs (scale, 0-3) that describes the severity for each affected organ/site at any given time and also measures functional impact.7 A global staging of severity (ie, mild, moderate, or severe) is derived by combining organ-specific scores, thereby replacing the “limited-extensive” nomenclature.7,17 The feasibility of using the NIH staging scale and the distribution of the individual organ scores and global severity stages has now been established in several large prospective studies.16,19,20 In the largest study of 298 cGVHD patients enrolled into the cGVHD consortium, it was determined that 10%, 59%, and 31% of patients had mild, moderate, or severe cGVHD, respectively.19 This new and practical scoring system enhances the quality and level of detail of cGVHD data recording and can be used in clinical practice or investigational trials (Figure 2).

Distribution of individual organ severity scores of cGVHD within global severity mild-moderate-severe staging categories. Data were obtained from the prospective study of the US cGVHD consortium (N = 298). The severity score accounts for both the magnitude of clinical manifestations and the degree of functional impairment. Reprinted with permission from Arai et al.19
Distribution of individual organ severity scores of cGVHD within global severity mild-moderate-severe staging categories. Data were obtained from the prospective study of the US cGVHD consortium (N = 298). The severity score accounts for both the magnitude of clinical manifestations and the degree of functional impairment. Reprinted with permission from Arai et al.19
Historically, several factors have been identified that predict the onset of aGVHD or cGVHD. However, in previous studies, aGVHD and cGVHD were generally referred to as disease that occurred within the first 100 days or after 100 days after transplantation. A recent large retrospective study of 2941 patients transplanted after myeloablative conditioning at the Fred Hutchinson Cancer Research Center evaluated risk factors for aGVHD and cGVHD using patients reclassified according to new NIH criteria.21 Risk factors for aGVHD grades II-IV included transplantation from HLA-matched unrelated donor (MUD) or a mismatched related or URD, use of total body irradiation (TBI) in the conditioning, and use of a female donor for a male recipient. Factors associated with lower risk of aGVHD were the use of rabbit antithymocyte globulin (ATG) in pretransplantation conditioning and chronic myeloid leukemia diagnosis. Grafting with growth-factor mobilized blood cells and patient/donor age were not associated with increased risk of grades II-IV aGVHD classified according to the NIH criteria. Risk factors for cGVHD scored by NIH criteria were similar to the aGVHD risk factors, with the exception of TBI in the conditioning. The use of growth factor–mobilized blood cells and donor or recipient age were also associated with cGVHD, suggesting that aGVHD and cGVHD are not entirely congruent processes. In a separate subanalysis, prior aGVHD grades III-IV were also associated with higher risk of cGVHD according to NIH guidelines.
Compared with these extensive data in the myeloablative setting, there is a relative paucity of data in patients receiving RIC. However, a recent study from the CIBMTR analyzed risk factors for classic aGVHD (within 100 days after transplantation) in a cohort of 5561 adult patients receiving transplantations between 1999 and 2005 (approximately 20% received allo-HCT after a RIC regimen).4 In the sibling donor cohort (n = 3191), the cumulative incidences of CIBMTR grades B-D and C-D aGVHD were 39% and 16%, respectively. In the URD cohort (n = 2370), the cumulative incidences of grades B-D and C-D aGVHD were 59% and 32%, respectively. Certainly, these data illustrate the magnitude of the aGVHD problem in a contemporary community–based cohort of patients. In an innovative way, this study analyzed the impact of the most common treatment packages currently used in transplantation protocols, because it took into account stem cell source, use of TBI, and conditioning intensity (Figure 3). A recent prospective study in 206 patients with cGVHD enrolled in an NIH natural history study identified TBI, especially in the RIC setting, as a significant prognostic factor for sclerotic-type cGVHD of the skin (Figure 4).22 Nevertheless, our current ability to predict aGVHD or cGVHD remains insufficiently reliable; however, it is possible that improved predictive criteria may be developed through integration of clinical and emerging biological markers.10,23

Cumulative incidence of aGVHD grade B-D in related donors (A; n = 3191) and URDs (B; n = 2370) stratified by treatment category. The analysis was performed through the CIBMTR. PBSC indicates peripheral blood stem cell. Reprinted with permission from Jagasia et al.4
Cumulative incidence of aGVHD grade B-D in related donors (A; n = 3191) and URDs (B; n = 2370) stratified by treatment category. The analysis was performed through the CIBMTR. PBSC indicates peripheral blood stem cell. Reprinted with permission from Jagasia et al.4

TBI is associated with an increased risk of development of sclerotic-type cGVHD. The association between TBI and sclerotic cGVHD was demonstrated most strongly among patients treated with RIC (P = .0114). Data are from the NIH study group prospective cohort. Reprinted with permission from Martires et al.22
TBI is associated with an increased risk of development of sclerotic-type cGVHD. The association between TBI and sclerotic cGVHD was demonstrated most strongly among patients treated with RIC (P = .0114). Data are from the NIH study group prospective cohort. Reprinted with permission from Martires et al.22
Prognostic factors for outcomes in patients with GVHD
The most established prognostic factors for poor survival and mortality in patients who develop aGVHD are grade III-IV severity and refractory disease.24–27 The characteristics most consistently associated with an increased risk of NRM among patients with cGVHD have been thrombocytopenia (< 100 × 109/L) and progressive onset of cGVHD from aGVHD. Several other factors associated with increased NRM in patients with cGVHD include: elevated bilirubin, poor Karnofsky performance status, steroid therapy at the time of onset, diarrhea, weight loss, GI involvement, HLA mismatch, increased patient age, prior aGVHD, and lack of therapeutic response to cGVHD treatment.18,28–33 Recently, a prognostic score has been developed for cGVHD that is defined by traditional criteria derived from a large cohort of 5343 patients reported to the CIBMTR between 1995 and 2004. The study cohort included patients of all ages treated by all graft sources, donor sources, and both myeloablative and RIC regimens. This analysis showed an OS for the whole cohort of 72% at 1 year and 55% at 5 years.5 The cumulative incidence of NRM was 21% at 1 year and 31% at 5 years; 6 risk groups were identified that had OS ranging from 15% to 90%.
It is important to emphasize that most studies evaluating prognostic factors for NRM and survival in GVHD are retrospective, from various treatment eras, include heterogeneous patient populations, and did not use contemporary diagnosis and staging criteria. However, in a positive vein, prospective data are now emerging in newly diagnosed and advanced patients due largely to the efforts of the cGVHD consortium in the United States and some single-center studies.19,34 These studies confirmed the significance of some previously recognized prognostic factors, such as low platelet count, progressive disease onset, and Karnofsky performance status, and also identified new prognostic factors such as NIH global severity stage (mild vs moderate vs severe), overlap syndrome, NIH lung score, and lymphopenia (Figure 5).15,16,19,20 Recent studies established the association between NIH mild-moderate-severe global stages and health-related quality of life.35 It is also expected that integration of established clinical prognostic factors and emerging biomarkers will assist in better individualization of GVHD therapy depending on the risk stratification.36,37

Cumulative incidence of OS according to NIH global severity at enrollment. Graph shows 2-year survival estimates, 95% confidence intervals (in parentheses), and hazard ratios (HR). Data are from the prospective study of the US cGVHD consortium (N = 298). Reprinted with permission from Arai et al.19
Cumulative incidence of OS according to NIH global severity at enrollment. Graph shows 2-year survival estimates, 95% confidence intervals (in parentheses), and hazard ratios (HR). Data are from the prospective study of the US cGVHD consortium (N = 298). Reprinted with permission from Arai et al.19
GVHD prophylaxis
aGVHD
The original aGVHD prophylaxis regimens developed during the 1970s used the folate antagonist methotrexate (MTX) due to its ability to delete proliferating donor lymphocytes. The initial MTX dosing regimen of days 1, 3, 6, and 11 and then once weekly through day 102 yielded an incidence of grades III-IV aGVHD of approximately 25%.38 Cyclosporine (CSA) entered clinical trials of GVHD prophylaxis in the late 1970s and showed equivalency with MTX in prospective studies.39 True progress in GVHD prevention occurred with combination regimens containing CSA and a short course of IV MTX (15 mg/m2 on day 1 and 10 mg/m2 on days 3, 6, and 11), which showed synergism and a survival benefit in BM transplantation from matched siblings and remains a commonly used regimen.4,40,41 No improvements in cGVHD incidence were seen with these regimens, again suggesting divergent pathogenic mechanisms. Attempts to improve outcomes by adding prednisone to the MTX/CSA combination did not yield positive results.42,43 During the 1990s, another calcineurin inhibitor, tacrolimus (TAC) used in combination with short-course MTX was tested in 2 large North-American phase 3 clinical trials after related and URD BM transplantation.44,45 Both trials showed reductions in overall aGVHD incidence (but not cGVHD) among patients receiving TAC/MTX relative to recipients of CSA/MTX; however, OS was not different. These studies prompted some centers to more frequently use the TAC combination, particularly in URD transplantations.
Mycophenolate mofetil (MMF), via its metabolite mycophenolic acid, inhibits proliferation of lymphocytes and is synergistic with calcineurin inhibitors in preventing GVHD. MMF also facilitates donor engraftment and is now widely used in RIC transplantations from related or URDs.38 Although GVHD prevention does not seem to be improved by use of MMF rather than MTX in calcineurin inhibitor–based regimens, there is a significant decrease in incidence and severity or oropharyngeal mucositis with the use of MMF.46,47
Although the combinations of calcineurin inhibitors and MTX or MMF have resulted in satisfactory rates of aGVHD and survival outcomes, these regimens are not uniformly effective and many patients are still dying from GVHD and related complications.4 Therefore, substantial efforts have been invested in attempts to improve on these calcineurin-inhibitor–based combinations. Anti–T-cell Abs have been explored as part of preparative regimens since the earliest days of allo-HCT; in uncontrolled studies, such Abs prevented GVHD but also increased risk of leukemia relapse, infections, non-relapse-related complications, and engraftment failures.48 Interpretation of these data is complicated by the huge variability in the studies, particularly in regard to the form of Ab used (at least 4 different forms have been used), source of the stem cell, type of donor, and conditioning regimen intensity. The best evidence for in vivo Ab efficacy is for ATG in URD BM transplantation after myeloablative conditioning.49 In a large randomized trial, patients who underwent allo-HCT from 8/8 MUDs (approximately 80% received peripheral blood stem cells) were randomly assigned to receive CSA/MTX with or without anti-Jurkat rabbit ATG. ATG recipients had significant reduction of grade II-IV and grade III-IV aGVHD from 51%-33% and from 24.5%-11.7%, respectively. ATG recipients also had a reduced 3-year incidence of extensive cGVHD (45.0% vs 12.2%).50 There was no statistically significant difference in relapse, NRM, mortality from infectious disease, or OS between groups. A smaller and older randomized study originally performed in the late 1990s showed similar short-term results.51 In addition, long-term follow-up showed reduced late pulmonary disease in the ATG arm, suggesting a potential long-term impact of in vivo ATG on health-related quality of life.52 Randomized trials to address the role of ATG, especially in cGVHD prevention, are progressing in the United States and Canada (www.clinicaltrials.gov identifiers NCT01295710 and NCT01217723, respectively). The role of ATG in RIC allo-HCT has not been formally tested because the success of these transplantations in terms of controlling relapse is more dependent on intact GVT reactions. A large retrospective CIBMTR study involving > 1400 patients confirm these concerns, because ATG recipients after RIC had increased risk of malignancy relapse, more NRM, more EBV lymphoproliferative disease, and lower OS and disease-free survival.53 Prospective randomized trials are needed to define the role of optimal dose and timing of ATG administration in the RIC allo-HCT setting.54,55 In a related approach, potent and practical techniques for ex vivo T-cell depletion strategies have been evaluated to prevent GVHD. Recently, a phase 2 study in acute myeloid leukemia patients in remission (mostly in in first complete remission [CR1]) demonstrated feasibility of such an approach in related donor transplantations using myeloablative conditioning devoid of posttransplantation systemic immunosuppression.56 In that study, the incidences of aGVHD and cGVHD were low and relapse did not appear to be increased; however, survival rates were not different from historical controls.
Another pharmacological approach to preventing GVHD has been developed by investigators at the Dana-Farber Cancer Institute through the use of sirolimus, an mTOR inhibitor, as an addition to TAC and MTX.57 In addition to effector T-cell inhibition, sirolimus can preserve regulatory T cells after transplantation, thereby adding to GVHD control. In a single-arm phase 2 study, the substitution of sirolimus for MTX in combination with TAC after myeloablative conditioning resulted in grade II-IV aGVHD of 20.5% and grade III-IV of 4.8%; no differences in outcomes were observed between recipients of related or URDs.58 This approach has been extended into the RIC setting, with results indicating that the addition of MTX to sirolimus and TAC is not necessary.59,60 These data support the utility of sirolimus as a second agent with TAC in GVHD prophylaxis. However, due to an increased risk of veno-occlusive disease, sirolimus should not be used with myeloablative doses of busulfan or in the TBI-based myeloablative regimens if combined with MTX.61
Because long-term administration of calcineurin inhibitors has toxicities and impairs T-cell development, Johns Hopkins University investigators are testing the use of high-dose posttransplantation cyclophosphamide (Cy) as sole prophylaxis for GVHD after HLA-matched related and URD T cell–replete BM transplantation.62 Cy, when given early after transplantation, acts similarly to MTX in terms of deleting rapidly dividing alloreactive T cells. Hematopoietic stem cells contain high levels of aldehyde dehydrogenase, which converts 4-hydroxycyclophosphamide into a nonalkylating metabolite, thus sparing stem cells from the antiproliferative activity of this agent. Cy was given at a dose of 50 mg/kg on days 3 and 4 after transplantation with myeloablative Bu-Cy conditioning without addition of any other systemic immunosuppression.62 The median time to neutrophil engraftment was 23 and 25 days in matched related donor (MRD) and URD patients, respectively, without use of exogenous colony stimulating factors; in addition, there was a relatively low treatment-related mortality of 13% and 21% at 2 years for MRD and MUD, respectively. Grade II-IV aGVHD incidence was 42% (MRD) and 46% (URD), with grade III and IV occurring in 12% and 8% of patients, respectively. Perhaps the most impressive clinical result of the Cy regimen was the low cumulative incidence of cGVHD, which was 10%. The potential advantage of this approach is selective elimination of host-reactive donor lymphocytes within days after transplantation, with relatively rapid recovery of other immunologic functions and without protracted exposure to calcineurin inhibitors (which may interfere with the induction of posttransplantation tolerance). Comparative prospective studies should define the risk of malignancy relapse after using the posttransplantation Cy approach.
Several additional agents are of potential interest or are in the early stages of clinical development for GVHD prevention.63 A phase 1/2 study demonstrated promise in aGVHD prevention using the proteasome inhibitor bortezomib in addition to TAC and low-dose MTX.64 A strikingly low incidence of gastrointestinal and liver aGVHD was observed in a recent phase 1/2 study using maraviroc, a well-tolerated oral CCR5 antagonist.65 Prospective studies for aGVHD prevention are summarized in Table 1.
Prospective studies of aGVHD prevention

P values are reported only in cases when the difference was statistically significant.
MSD indicates HLA matched sibling donor; BMT, BM transplantation; AA, aplastic anemia; MAC, myeloablative conditioning; PDN, prednisone or equivalent dose of another corticosteroid (most commonly, IV methylprednisolone); SIRO, sirolimus; mm, HLA mismatch; and IS, immunosuppression.
cGVHD
Due to a relatively limited understanding of cGVHD pathogenesis, developing preventive strategies specifically for this complication of allo-HCT has been more difficult. None of the current calcineurin-inhibitor–based pharmacological approaches that successfully prevent aGVHD has a major impact on cGVHD (Table 1). Whether the development of calcineurin-inhibitor–free aGVHD prevention methods would have better impact on cGVHD remains to be determined.66 Although cGVHD can be reduced by methods that quantitatively eliminate donor T cells by in vivo or ex vivo T-cell depletion or posttransplantation high-dose Cy, the success of such approaches is constrained by a higher rate of malignancy relapse or infections, thereby leading to lack of a survival advantage or even reduced survival.48,53 Two randomized trials were specifically designed to prevent cGVHD with study drugs added to the standard calcineurin inhibitor–based aGVHD prevention. One placebo-controlled randomized trial tested hydroxychloroquine administered on days 21-365 after transplantation in 95 patients and did not show effects on aGVHD or cGVHD or on survival.67 The other randomized placebo-controlled trial that tested thalidomide in 59 patients showed detrimental effects due to an increased rate of cGVHD flares and impaired survival in the experimental arm.68 The reasons for the discrepancy in cGVHD prevention between pharmacological suppression and T-cell depletion strategies are not clear, and a better understanding of these processes may aid in the development of new treatments to prevent cGVHD and hopefully harness GVT effects. Based on the recent information about the role of B cells in cGVHD, anti-CD20 Ab strategies are currently being tested for their potential in preventing cGVHD.69
GVHD treatment
aGVHD frontline therapy
The current model of aGVHD development includes 3 phases: (1) tissue damage induced by the preparative regimen; (2) priming and activation of donor T cells, with CD8 T cells being stimulated by residual host APCs and CD4 T cells being stimulated by donor APCs presenting host-derived antigens; and (3) target tissue damage induced directly by cytotoxic T cells and indirectly by inflammatory cytokines.6 Therefore, modulation of donor-alloreactive effector T cells remains a major focus of current therapies for aGVHD. Nonetheless, other cell populations including regulatory T cells, dendritic cells, natural killer T cells, and B cells or other mechanisms such as modulation of tissue or vascular endothelium damage signals may represent future targets for novel approaches to GVHD therapy.63 Although several agents have been tested over the last 30 years for the systemic therapy of aGVHD, no product is approved by the US Food and Drug Administration (FDA) for use in aGVHD or cGVHD. Poor standardization of GVHD clinical studies has been identified as a major obstacle to the progress of clinical trials and the development of new treatments.70 Recent recommendations of the American Society of Blood and Marrow Transplantation (ASBMT) recognize this fact and raise the bar required for study implementation and publications of future studies in aGVHD.27
Patients with aGVHD requiring systemic therapy (grades II-IV) are typically started on 2 mg/kg/d (divided in 2 doses) of methylprednisolone or a prednisone equivalent. Concurrently, patients are usually continued on calcineurin-inhibitor–based GVHD prophylaxis. There are strong data from an Italian randomized trial showing no advantage of initial treatment with doses higher than 2 mg//kg/d of methylprednisolone.71 An important goal of GVHD therapy is to minimize complications associated with high-dose glucocorticoids. Some centers have attempted to begin treatment with methylprednisolone-equivalent doses of 1 mg/kg/d in patients with milder (grade II) presentations of aGVHD with dose escalation to 2 mg/kg/d if patients exhibit progressing GVHD after 3 days of therapy. In a large retrospective study of 733 patients, this approach showed no adverse impact on outcomes and yielded a reduction of total steroid dose by approximately 50%. Although promising, this strategy needs to be validated prospectively.72 In one randomized trial, a successful strategy to reduce systemic glucocorticoid exposure consisted of the incorporation of potent topically acting, poorly absorbable glucocorticoid beclomethasone dipropionate into therapy for GI GVHD in patients who had milder forms of grade II aGVHD (ie, diarrhea < 1000cc, skin body surface area < 50%, and no liver involvement).73
The optimal rate of tapering steroid doses after initial treatment has not been defined. One randomized trial showed no advantage of a prolonged taper schedule of prednisone.74 In general, tapering of steroids should begin when aGVHD manifestations start showing major improvement. Tapering typically continues at 10% of the starting dose every 3-5 days, slower after prednisone doses are decreased to less than 20-30 mg/d.27
Because only approximately 60% of aGVHD patients respond to systemic steroids and many of these responses are not durable, attempts have been made to evaluate other agents in addition to prednisone compared with prednisone alone for initial therapy.27,75 Agents evaluated in prospective studies have included Abs against IL-2R, high-dose steroids, horse ATG, anti-TNF drugs, MMF, pentostatin, and sirolimus (Table 2).76–82 In most cases, the second agent yielded no benefit, and in one study the additional intervention was detrimental (daclizumab). In a Blood and Marrow Transplant Clinical Trials Network (BMT-CTN) 4-arm randomized phase-2 trial, the addition of MMF to methylprednisolone showed the most promise for initial therapy of aGVHD compared with etanercept, denileukin, or pentostatin.81 A definitive phase 3 BMT-CTN trial evaluating corticosteroids with MMF versus placebo as initial treatment for aGVHD has been recently closed for accrual and the data analysis is being awaited (www.clinicaltrials.gov identifier NCT01002742).
Selected studies of aGVHD frontline therapy
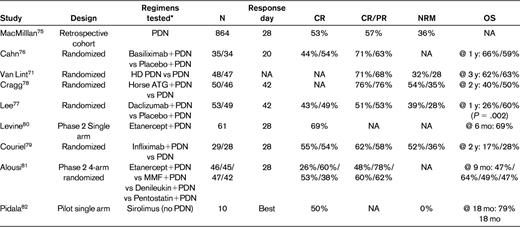
Meeting quality criteria per American Society for Blood and Marrow Transplantation recommendations.27
PDN indicates prednisone or equivalent dose of another corticosteroid; NA, not available; and HD PDN, high-dose prednisone or equivalent.
*All typically administered in addition to a calcineurin inhibitor.
For better design of future studies, it would be invaluable to develop methods to distinguish the patient population that is likely to do poorly with frontline steroids. The abovementioned 4-arm BMT-CTN randomized phase 2 trial provided a platform for recent proteomics discovery and validation of 6 aGVHD biomarkers that predict response and survival after frontline therapy. These data should aid in the design of studies and may be used for early risk stratification in combination with known clinical prognostic factors.36 The time point for therapy response assessment should be standardized across trials because the response rate typically increases over time. Consensus has emerged to use day 28 as a standard response evaluation time point in frontline aGVHD treatment studies because this end point is also correlated with NRM.25,75
aGVHD second-line therapy
If aGVHD progresses within 3 days or is not improved after 5-7 days of initial treatment with standard-dose methylprednisolone, then the GVHD is considered to be steroid refractory and second-line therapy is warranted. Sometimes, if aGVHD is of a milder grade II, a longer observation interval of up to 2 weeks is acceptable. Second-line treatments of aGVHD are associated with significant toxicities, high failure rates, and 1-year survival rates of approximately 20%-30%.27,83,84 Very few prospective studies have evaluated second-line therapy for aGVHD, and interpretation of these studies is hampered by the lack of standardization, the absence of validated evaluation end points (day-28 response is not validated for second-line therapy), and small numbers of patients. This topic has been extensively and critically reviewed in the recently published recommendations of the ASBMT.27 Agents that have been investigated over the last 2 decades in these trials include: low-dose MTX, MMF, extracorporeal photopheresis, IL-2R targeting (ie, basiliximab, daclizumab, denileukin and diftitox), alemtuzumab, horse ATG, etanercept, infliximab, and sirolimus.27 On average, approximately 50% of patients responded (CR or partial remission) to second-line therapy, the median survival was approximately 6 months, and there was no evidence that any specific agent was either preferred or particularly counterproductive. Because comparative data on superior efficacy for any particular agent does not exist, the choice of a second-line regimen is made based on the effects of prior treatments, desired toxicity profile, considerations for drug interactions, logistical practicality, costs, and patient and physician preferences. Second-line treatments, especially those associated with depression of T cells, are associated with increased infection and viral reactivation (including CMV, EBV, HHV-6, adenovirus, and polyoma); early tapering of corticosteroids may be considered to minimize infection risk.85 Development of treatments that rely less on steroids or immune depletion would be highly beneficial.63 Retrospective studies in large single-center or registry cohorts should also be performed on contemporary patients to determine whether the overall results from published studies are truly representative of broader practices. Such contemporary studies are lacking.
cGVHD frontline therapy
The typical course of cGVHD is protracted, lasting on average 2-3 years. Ultimately, approximately 85% of patients who survive beyond 5 years after diagnosis are able to discontinue systemic therapy.32 The goal of therapy of cGVHD is to stop the destructive immunological process, alleviate symptoms, and prevent disease progression that may lead to irreversible disability or death. The ultimate goal is to establish immunological tolerance and withdraw immunosuppressive therapy. These patients require continued monitoring for late effects of transplantation and cancer therapy.86 Principal components of cGVHD therapy include systemic treatment with immunosuppressants or immune modulators integrated with ancillary therapy and supportive care.87–90 Systemic therapy should be considered for patients who meet criteria for moderate to severe global severity (involvement of 3 or more organs or with an NIH score of 2 or greater in any single organ or any lung involvement).7 Some experts incorporate the presence or absence of published high-risk features (eg, thrombocytopenia or progressive onset) and the underlying reason for transplantation (eg, malignant vs nonmalignant disease) or current comorbid conditions (eg, infection) into decisions of whether to treat with systemic immunosuppression.
Current results of initial therapy of cGVHD are still suboptimal and the standard of care recommendation is to enroll patients in clinical trials if such a trial is available. The most widely used initial systemic treatment of cGVHD relies on prednisone in conjunction with a calcineurin inhibitor.87,91–95 For those patients who have not responded by the 3-month time point or who progress, alternative salvage regimens should be instituted.
Attempts to improve outcomes of primary therapy in cGVHD continue, although very few studies have addressed this issue. Two randomized trials of thalidomide as part of initial therapy yielded no clinical benefit of the addition of this agent to standard CSA and prednisone.96,97 Martin et al reported results of a double-blind, randomized multicenter trial in 151 patients to determine whether the addition of MMF improves the efficacy of initial systemic treatment.98 The trial was closed prematurely before the target accrual of 230 patients due to a low probability of reaching the primary end point of 2-year survival off of systemic immunosuppression without resorting to secondary treatment, indicating that MMF should not be added to the initial treatment of cGVHD. The Children's Oncology Group (COG) recently published the results of a multicenter double-blind, placebo-controlled phase 3 trial evaluating hydroxychloroquine added to standard therapy with steroids and calcineurin inhibitor for children with newly diagnosed cGVHD.99 The primary end point of the trial, CR at 9 months of therapy, was achieved in 28% of patients in the experimental arm versus 33% in the placebo arm. The CR rates in patients with moderate versus severe cGVHD per NIH criteria were 44% and 15%, respectively. That study, which also incorporated extensive immunology testing to evaluate the pathophysiology of cGVHD,37 closed prematurely for slow accrual; however, it provided important information for the future design of trials in pediatric patients. Six randomized phase 3 studies of cGVHD initial therapy are summarized in Table 3.
Randomized phase 3 studies of cGVHD initial therapy
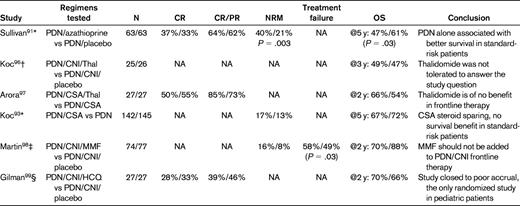
PDN indicates prednisone; CNI, calcineurin inhibitor; Thal, thalidomide; HCQ, hydroxychloroquine; and NA, not available.
*All randomized patients had platelet count > 100 000/μL.
†All patients had high-risk cGVHD (ie, thrombocytopenia and progressive onset).
‡Multicenter trial (treatment failure was defined as the initiation of secondary therapy or bronchiolitis obliterans or recurrent malignancy or death from causes other than recurrent malignancy).
§COG multicenter trial.
cGVHD second-line therapy
Approximately 50% of patients with cGVHD fail to achieve control after initial therapy with steroids by 1 year after diagnosis.30,100 Therefore, there is a frequent need for the initiation of secondary treatments.90 Indications for secondary treatment include: failure of primary steroid-based therapy, failure of any subsequent line of therapy as manifested by progression of cGVHD, stable persistence of cGVHD despite 4-12 weeks of sustained therapy or inability to taper immunosuppression without recurrence of clinical manifestations. Earlier initiation of secondary therapy would be appropriate in patients with more severe cGVHD, whereas a longer trial of initial therapy would be appropriate in patients with sclerotic skin changes or other slowly reversible manifestations of cGVHD. Inability to tolerate therapy (eg, steroid myopathy or calcineurin inhibitor– and/or sirolimus-induced thrombotic microangiopathy) may also be considered an indication for secondary treatment.
There is currently no standard of care for cGVHD patients who fail frontline steroid-based therapy, so enrollment in a clinical trial is recommended. Several (mostly smaller) phase 2 studies of secondary or salvage regimens have been published, and most of them report response rates of 25%-80% and 1- to 3-year survival rates of approximately 70%.90,101 Responses are frequently incomplete and not durable. Recently, the German-Austrian-Swiss cGVHD study group organized an international project and provided evidence-based guidelines for clinical practice in cGVHD secondary therapy.90 Agents to be considered for secondary therapy are shown in Table 4. All recommendations beyond steroids are grade C (optional) and most levels of evidence are level III (expert opinion), with the exception of extracorporeal photopheresis, pentostatin, rituximab, and thalidomide recommendations, which are assessed as level II (nonrandomized studies). Because there is no therapeutic standard, choices in salvage therapy for cGVHD are made based on other factors, including patient history of previous treatments, choosing agents without overlapping toxicities, patient and physician personal preference, logistics, and costs. In patients with advanced cGVHD, it is important to maintain awareness of the possibility that some symptoms may be due to irreversible organ damage and not to active disease.
Recommendations for second-line systemic treatments in cGVHD and levels of evidence per the Regensburg Consensus Conference90
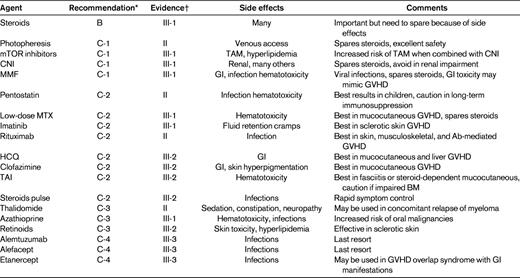
TAM indicates transplantation-associated microangiopathy; CNI, calcineurin inhibitor; HCQ, hydroxychloroquine; and TAI, total abdominal irradiation.
*Recommendations were indicated as follows: A, should always be offered; B, should generally be offered; C, optional; C-1, use in second-line therapy justified; C-2, use in greater than second-line therapy justified; C-3, use limited to specific circumstances because of increased risk profile; C-4, experimental; and D, should generally not be offered.
†Evidence was indicated as follows: I, evidence from > 1 randomized controlled trials; II, evidence from 1 well-designed clinical trial without randomization, from cohort analytic studies, or from dramatic results of uncontrolled experiments; III, evidence from opinions of respected authorities; III-1, several reports from retrospective evaluations or small uncontrolled clinical trials; III-2, only 1 report from small uncontrolled clinical trial or retrospective evaluations; and III-3, case reports.
There is clearly a tendency in recent years toward developing more rationally based targeted immunomodulating therapies for steroid-refractory cGVHD that are based on our emerging understanding of cGVHD pathophysiology.102,103 Such examples include recent promising uses of low-dose IL-2 for stimulation of T-regulatory cells,104 imatinib for PDGF pathway inhibition,105 and leukotriene inhibitors106 or rituximab for B-cell targeting.107 As in aGVHD, there is an urgent need for better standardized clinical trials of new agents to advance therapeutic success in cGVHD.70 The NIH consensus conference proposed a panel of cGVHD response measures to accelerate the development of short-term end points in clinical trials for drug development.108 These measures are currently being validated and refined in clinical studies.104,109–114 A very popular Blood journal Web site video-based teaching tool walks the viewer through a practical 18-minute clinical examination developed for training of health care providers who are involved in cGVHD clinical trials.115
Conclusions
In conclusion, are we making progress in GVHD prophylaxis and treatment? Randomized studies over last 3 decades established CSA- or TAC-based 2-drug combinations with MTX as standard prevention for aGVHD. Although the addition of ATG decreases both aGVHD and cGVHD, this strategy has not been shown to improve survival in patients with hematologic malignancy. More recent single-arm studies using new drugs added to calcineurin inhibitors show lower rates of aGVHD, but these findings must be confirmed in randomized trials. The calcineurin-inhibitor–free strategy of using posttransplantation Cy for selective removal of alloreactive T cells is a potential breakthrough, because it brings the possibility for fewer non-relapse-related complications and immunosuppression-free immunological reconstitution. The recent realization of transplantation treatment packages affecting the risk for aGVHD gives additional tools to the clinician when choosing the most adequate protocol for specific patients.4 There is currently no standard of care for pharmacological methods for the prevention of cGVHD; clinical trials in the myeloablative and RIC setting need to evaluate the role that ATG or posttransplantation Cy may play in cGVHD prevention in patients with hematologic malignancy. Because it is clear that the use of stem cells from the peripheral blood results in more cGVHD relative to BM grafts, clinicians today have the ability to select the most appropriate stem cell source for a particular patient.
Steroids are established frontline therapy for aGVHD, and there is no value of adding other drugs to steroids as part of frontline therapy. Better predictive schemes are needed to appropriately select patients for such studies. The most progress in aGVHD is emerging from an increased ability to identify novel biomarkers that may be combined with clinical prognostic factors for better individualization of patient therapy and stratification in clinical trials of new interventions. There is no evidence of any agent having an advantage for second-line treatment of aGVHD, and survival of these patients remains poor. Furthermore, larger-scale data in more recent patients are lacking. Steroids also remain standard frontline therapy for cGVHD, because none of the randomized studies in this setting demonstrated a value of adding another agent. In this setting, improved methods are required to identify patients at risk of failing steroids. There is no standard second-line therapy for cGVHD and no agent can be endorsed over the other. There is a need for better standardization of trials both in aGVHD and cGVHD.25,27,70 The NIH conference provided a set of definitions that are improving the standardization of clinical studies and increasing our awareness and understanding of the significance of the diverse clinical presentations of cGVHD.116 One major breakthrough in the cGVHD field has been the establishment of a national consortium that now enables investigators to effectively conduct multicenter observational and interventional studies.34 In addition, higher-quality prospective data are emerging from other national and international centers and groups studying cGVHD.
In summary, both aGVHD and cGVHD remain serious barriers to successful allo-HCT and it is not clear that a major improvement has occurred in our ability to prevent or treat these patients or that outcomes have improved over the last 20 years. Nevertheless, there is an increasing understanding of the biology, clinical manifestations, and transplantation-related factors, and the field is getting better organized. These advances will almost certainly lead to major progress in the near future. As the long list of new potential targets and respective drugs are identified, systems need to be developed for rapid testing of them in clinical practice. The current reality is that no single agent has yet been approved by the FDA for GVHD prevention or therapy. Although a primary goal of these efforts is to develop better therapies for GVHD, the ultimate goal is to develop treatments that will lead to effective prevention or preemption of life-threatening and disabling GVHD manifestations.
Acknowledgments
The authors thank our many collaborators and colleagues over the years and the colleagues, nurses, and clinical staff at the Experimental Transplantation and Immunology Branch and the NCI who make our work possible.
This work is supported by the Center for Cancer Research, National Cancer Institute and National Institutes of Health. Statements included in this article do not represent the official position of the National Cancer Institute, the National Institutes of Health, or the US Government.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: No off-label use of any specific agent; however, because there are no FDA-approved standard therapies for GVHD, any medication mentioned in this context will be, strictly speaking, “off-label.”
Correspondence
Steven Z. Pavletic, MD, Graft-versus-Host and Autoimmunity Unit, Experimental Transplantation and Immunology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bldg 10, Rm CRC-3-3330, 10 Center Dr, Bethesda, MD 20892; Phone: 301-402-4899; Fax: 301-480-4354; e-mail: pavletis@mail.nih.gov.
