
Abstract
Sickle cell disease and thalassemia have distinctly different mutations, but both share common complications from a chronic vasculopathy. In the past, fetal hemoglobin–modulating drugs have been the main focus of new therapy, but the increased understanding of the complex pathophysiology of these diseases has led to the development of novel agents targeting multiple pathways that cause vascular injury. This review explores the pathophysiology of hemoglobinopathies and novel drugs that have reached phase 1 and 2 clinical trials. Therapies that alter cellular adhesion to endothelium, inflammation, nitric oxide dysregulation, oxidative injury, altered iron metabolism, and hematopoiesis will be highlighted. To evaluate these therapies optimally, recommendations for improving clinical trial design in hemoglobinopathies are discussed.
Introduction
Sickle cell disease (SCD) and thalassemia have distinctly different mutations, but both share common complications from a chronic vasculopathy.1–4 In SCD, polymer formation leads to membrane destruction and hemolysis, which initiates a cascade resulting in a vasculopathy characterized by hypoxia reperfusion injury.5 Thalassemia syndromes result from consequences of unbalanced globin chain synthesis associated with an increased redox activity of the abnormal hemoglobin, resulting in membrane damage and hemolysis.1,6 The development of drugs to increase fetal hemoglobin has been the major therapeutic strategy in the treatment of both disorders. Although new fetal hemoglobin–modulating agents are being studied, only hydroxyurea has shown long-term benefit. Recently, novel agents targeting the multiple pathways causing vascular injury in hemoglobinopathies have been developed.7
Endothelial damage
Endothelial damage is central to the pathophysiology of hemoglobinopathies.8 The initial step in the cascade of events is injury to the RBC membrane, exposing phosphatidylserine and releasing hemoglobin.9,10 This results in nitric oxide (NO) deficiency, activation of several mechanisms that lead to increased adherence to the endothelium, and impaired blood flow.2 This repeated process leads to ischemia reperfusion injury, which is accompanied by a marked increase in cytokines and activation of leukocytes, procoagulants, and adhesion molecules.11 Simultaneously, cytoprotective mediators such as antioxidants are depleted. Increased adhesion of cells to the vascular endothelium, inflammation, NO dysregulation, oxidative injury, and altered iron metabolism are discussed in the first section. This is followed by a section on therapeutic agents targeting these pathways.
Adhesion
Increased adhesion of sickle cells to the endothelium is a key step in sickle vasculopathy.8,12,13 Damaged sickle cells adhere to the endothelium by adhesion molecules expressed on both the RBC and the endothelium. The RBC has the receptors VLA-4/α4β1 integrin, CD36, and integrin-associated proteins (IAP/CD47). These receptors bind to endothelial ligands such as E-selectin, P-selectin, αvβ3 integrin, and VCAM-1, either directly or via bridging molecules such as thrombospondin and VWF.14
Inflammation
Inflammation is a central factor in the severity of the sickle vasculopathy.7,8,15–18 Stable SCD patients have chronic elevation in proinflammatory cytokines, including C-reactive protein, TNF, IL-1, IL-8. Endothelial production of TNF-alpha and IL1-beta are induced by sickle cells. Monocytes are activated in SCD and enhance inflammation and the production of reactive oxygen species (ROS). Activated monocytes demonstrate differential expression of genes involved in inflammation and heme metabolism.19 NF-κB/p65, Kruppel-like factor 2 (KLF2), and other transcription factors that regulate pathways of inflammation are increased in SCD.
NO
NO, a critical vasodilator, is synthesized by endothelial cells from its obligate substrate, L-arginine. NO maintains vascular tone and reactivity and is depleted in both SCD and thalassemia.2,3,20 Hemolysis releases hemoglobin into the plasma, which destroys NO and forms ROS.10,21 Vasodilation is inhibited and inflammatory, adhesion, and procoagulant molecules are increased. NO depletion has additional protean effects, including the production of endothelin-1, a strong vasoconstrictor. Erythrocyte arginase released during hemolysis redirects arginine metabolism away from NO and induces aberrant vessel wall remodeling. Hemoglobinopathy patients are arginine deficient and demonstrate increased arginase activity. Patients have elevated asymmetric dimethylarginine, which inhibits arginine transport, promotes endothelial dysfunction, and is a risk factor for early mortality.20,22
Oxidative injury
Oxidative stress damages the RBC membrane and the vascular endothelium.23–26 The production of ROS is significantly elevated in hemoglobinopathies compared with normal controls. Denaturation of hemoglobin releases iron, which increases ROS, resulting in lipid and protein oxidation and mitochondrial dysfunction. Repeated ischemic reperfusion also markedly increases ROS and generates superoxide (O2) peroxynitrate (ONOO−). Continued oxidative stress induces an inflammatory response with cellular injury, apoptosis, and activation of the NF-κB and AP-1 pathways. The antioxidant defense system is impaired. Patients have decreased levels of glutathione, superoxide dismutase, heme oxygenase-1, lipoic acid, catalase, and other antioxidants.27
Iron metabolism and erythropoiesis
Both thalassemia and SCD patients undergo iron-induced injury and have altered iron metabolism.28–30 Intriguing data suggest that these iron-overloaded patients have iron-limited erythropoiesis and hyperactivation of Jak-2 kinase. In the thalassemia mouse model, transferrin injections increase hemoglobin, decrease ineffective erythropoiesis, and improve iron distribution. In other diseases associated with iron injury, human transferrin injections are protective, highlighting the potential therapeutic role of transferrin. When treated with Jak-2 inhibitors, ineffective erythropoiesis and extramedullary hematopoiesis decrease. Recognition of the role of hepcidin and ferroportin in iron metabolism has led to the development of novel hepcidin agonists and antagonists that improve erythropoiesis and decrease hemosiderosis.
Novel therapies (Table 1)
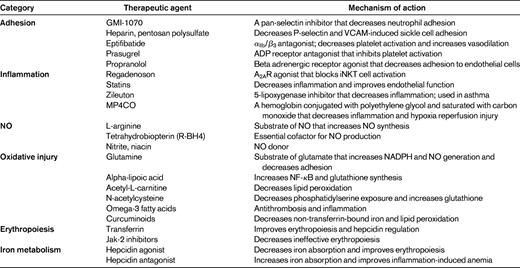
Adhesion
Phase 1 and 2 trials evaluating inhibitors of cellular adhesion are being conducted. Infusion of GMI-1070, a pan-selectin inhibitor, results in improved flow secondary to decreased neutrophil adhesion by its inhibition of E-selectin and neutrophil activation.31,32 A clinical trial to determine its efficacy in the treatment of acute vasoocclusive crisis is near completion (www.clinicaltrials.gov identifier NCT00911495). Endothelial P-selectin plays a central role in the impaired microvascular blood flow observed in SCD. Heparin decreases sickle cell adhesion through P-selectin.33 Computer-assisted intravital microscopy demonstrated increased blood flow after heparin treatment. Clinical trials with low-molecular-weight heparin decreased vasoocclusive events in SCD patients.34 Recently, a small phase 2 trial using a sulfated polysaccharide similar to heparin but with fewer anticoagulant properties was reported.35 There was a significant decrease in plasma VCAM-1 with no changes in daily pain scores. The importance of activated platelets on the vasculopathy of SCD have led to clinical trials with antiplatelet therapy.14 Clinical trials of eptifibatide, an αIIb/β3 antagonist, demonstrated decreased platelet activation and increased vasodilation in SCD patients.36 A controlled phase 2 trial evaluating its effects on vasoocclusive crisis and bleeding has been initiated. Prasugrel is an ADP receptor antagonist that is US Food and Drug Administration (FDA) approved for the use in acute coronary syndromes. In randomized trials in SCD, platelet activation was prevented and preliminary results suggest a decrease in pain rate.37 A randomized trial evaluating the inhibitory effects of propranolol, a beta-adrenergic receptor antagonist, on soluble markers of endothelial adhesion and activation in SCD has been initiated (www.clinicaltrials.gov identifier NCT01077921). This trial is based on preliminary data indicating that propranolol can safely abrogate epinephrine-stimulated adhesion of sickle cells to the endothelium.
Inflammation
SCD is a chronic inflammatory illness that sustains and magnifies vasoocclusion and the sickle vasculopathy. SCD patients have increased numbers and activation of invariant natural killer T (iNKT) cells compared with normal controls. A2A agonists are highly effective at reducing ischemic reperfusion injury by limiting the inflammatory response.38 Their primary benefit is their effect on iNKT cells. In the SCD animal model, A2a agonists prevent or minimize acute sickle lung injury.39 A phase I safety study of regadenoson (a selective A2AR agonist approved for myocardial perfusion studies) in patients with SCD was completed safely, and finding of decreased iNKT cells has led to a multicenter efficacy study. In the SCD mouse, statins protect from hypoxia reperfusion injury and have a direct inflammatory effect. In SCD patients, statins decrease inflammation (including C-reactive protein and IL-6 levels) and increase plasma NO.40 SCD patients have elevated levels of 5-lipoxygenase, a potent inflammatory leukotriene. Zileuton, a specific inhibitor of 5-lipoxygenase, is FDA approved for asthma. Beneficial effects in the SCD animal model have led to a phase 1 trial in SCD (www.clinicaltrials.gov identifier NCT01136941). Carbon monoxide inhibits inflammation and hypoxia-induced vasoocclusion in transgenic mice.41 MP4CO, a human hemoglobin conjugated with polyethylene glycol and saturated with carbon monoxide, protects transgenic mice from hypoxia reperfusion injury and has anti-inflammatory properties. This observation has led to a proposed 32-patient safety trial in SCD (www.clinicaltrials.gov identifier NCT01356485).10
NO-arginine dysregulation
Dysregulation of NO metabolism is a common denominator in the pathogenesis of hemoglobinopathy vasculopathy. Inhaled NO in SCD has resulted in variable clinical responses. A recent phase 2 trial for adult SCD patients treated with NO for painful crises showed no benefit.42 In SCD mouse models, arginine supplementation inhibits RBC Gardos channels, reduces RBC density, improves perfusion, and decreases vasculopathy and mortality. In SCD patients with pulmonary hypertension, arginine supplementation increases plasma NO and rapidly decreases pulmonary artery pressure by 15%.43 Arginine's effect on NO is dose dependent. To overcome the impact of arginase and asymmetric dimethylarginine on arginine bioavailability, high doses are necessary. Low-dose arginine has little impact on NO synthesis, as confirmed in a prophylactic arginine trial in SCD.44 A recent randomized, double-blind, placebo-controlled study of high-dose arginine supplementation in hospitalized SCD patients with vasoocclusive crisis was completed45 and found a > 56% reduction in narcotic use in patients receiving arginine compared with controls. These observations have led to a multicenter trial with arginine supplementation for the treatment of acute painful crisis. Other approaches to increasing NO bioavailability include phase 1 and 2 clinical trials evaluating sodium nitrite, niacin, and tetrahydrobiopterin (R-BH4; www.clinicaltrials.gov identifier NCT01033227, NCT00508989).46
Oxidative injury
Oxidative injury is an important feature of both SCD and thalassemia.26,27,47 Oxidative stress is increased by iron overload, membrane damage, and the depletion of the natural antioxidant defense mechanisms that characterize hemoglobinopathies. Reduced glutathione is the primary thiol redox buffer in RBCs and is important in maintaining vascular integrity.48 Glutamine is metabolized to glutamate, the glutathione precursor, and preserves intracellular nicotinamide adenine dinucleotide, which is necessary for glutathione recycling. Oral supplementation of glutamine in SCD increases the NAD redox potential and may improve sickle erythrocyte adhesiveness.49 Oral supplementation also decreases the resting energy expenditure in children with SCD. A multicenter phase 3 trial of glutamine supplementation to prevent vasoocclusive crisis is ongoing. Glutamine depletion in the RBC has been associated with elevated tricuspid regurgitant jet velocity and markers of hemolysis and oxidative stress.48 These observations have led to a phase 2 trial of oral glutamine therapy for hemolysis-associated pulmonary hypertension in SCD and thalassemia (www.clinicaltrials.gov identifier NCT01048905). Alpha-lipoic acid augments cellular stress response by increasing the transcription of antioxidant genes, decreasing NF-κB, and increasing glutathione synthesis. It is now in pilot studies in hemoglobinopathies. Acetyl-l-carnitine is an essential nutrient that facilitates the entry of long-chain fatty acids into the mitochondria and decreases lipid peroxidation in tissue. Recent trials in thalassemia suggest a benefit in cardiac function.50 In pilot studies, N-acetylcysteine decreases markers of oxidative injury in SCD, including phosphatidylserine exposure and glutathione levels.51 These observations have led to recent combination trials of antioxidants. Alpha-lipoic acid and acetyl-L-carnitine have a synergistic antioxidant effect52 and a phase 2 trial is evaluating their efficacy in decreasing vasoocclusive events. Pilot studies have demonstrated that omega-3 fatty acids decrease vasoocclusive events and plasma markers of thrombosis.53 These findings have led to new trials evaluating their benefit for inflammation and SCD (www.clinicaltrials.gov identifier NCT01202812). Recently, curcuminoids have been found to decrease non-transferrin-bound iron, inhibit lipid peroxidation, and lower malondialdehyde levels. In iron-overloaded animal models, curcuminoids decreased heart iron concentrations.54 These observations have led to interest in human trials.
Iron metabolism and erythropoiesis
Increased understanding of the regulation of erythropoiesis and iron metabolism has led to several potential therapeutic options.29,55 Ineffective erythropoiesis and anemia in thalassemia appear to be, in part, secondary to a functional iron deficiency caused by inadequate circulating transferrin. Transferrin injections in animal thalassemia models improve effective erythropoiesis, improve hepcidin expression, and decrease labile plasma iron levels.30,56 Ineffective erythropoiesis can also be improved by Jak-2 inhibitors. Increased Jak-2 levels limit erythroid differentiation and lead to ineffective erythropoiesis.28 In animal models, Jak-2 inhibitors reverse splenomegaly and improve erythropoiesis. The increased understanding of hepcidin ferroportin system has led to the development of several hepcidin agonists that decrease iron overload and may improve erythropoiesis. Concomitantly, there has been development of hepcidin antagonists to treat iron-restrictive anemias. These antihepcidin therapies have been developed commercially and will be entering human trials.
Clinical trial design: an area of potential improvement
There is a dramatic increase in potential novel therapy for hemoglobinopathies that are based on improved understanding of the pathophysiology and vasculopathy of these diseases. The clinical trial design needs to be improved to uncover the clinical significance of these therapies in these relatively rare diseases.57,58 End points needs to be reassessed and expanded, including functional and quality-of-life outcome measures.59
Previous hemoglobinopathy studies were plagued by inadequate enrollment; for example, SCD trials evaluating dexamethasone60 and transfusion therapy61 for acute chest syndrome. Study design addressing enrollment requirements realistically needs to be implemented.57,58 Clinical trials that incorporate the physiologic basis for different SCD phenotypes may improve study success. Therapies for the high-hemoglobin phenotypes characterized by repeated acute events may be different from the phenotypes characterized by low hemoglobin, limited acute events, and chronic organ disease. Novel agents may cause clinically significant benefits in a subgroup of patients, but not be further developed because of the chosen end points. Senicapoc, a Gardos channel inhibitor, increases hemoglobin and would be beneficial in a subgroup of very anemic patients, but clinical trials were abandoned because it did not sufficiently decrease vasoocclusive events. The phase 3 trial evaluating sildenafil (which increases NO signaling pathway) used painful events as a stopping point for the study. Sildenafil may enhance pain sensitivity, but its beneficial effect on vasculopathy and CNS injury are missed when acute painful events are a primary or stopping end point.62 Multimodality therapies designed to address different biological pathways responsible for the pathology of hemoglobinopathies are necessary to develop optimal therapy for SCD. In oncology, combination therapy has changed the prognosis of many cancers dramatically, but in SCD we rely on the hope of a single magic bullet. This approach needs to be reassessed.
In summary, there are reasons for optimism in the treatment of SCD. New therapies offer major improvements. This growth in our understanding of the pathophysiology of the disease should be accompanied by a more sophisticated approach to clinical trials.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Drugs described in research studies are not being recommended for clinical off-label use; the discussion focuses on their experimental effects on the pathophysiology of hemoglobinopathies.
Correspondence
Elliott Vichinsky, MD, Hematology/Oncology, Children's Hospital and Research Center Oakland, 747 52nd St, Oakland, CA 94609; Phone: 510-428-3651; Fax: 510-450-5647; e-mail: evichinsky@mail.cho.org.
