
Abstract
Traditionally, cytopenias are classified as deficiency mediated, immune mediated, BM failure induced, renal, or idiopathic, with the latter including the so-called idiopathic cytopenias of undetermined significance (ICUS). Clinical findings, symptoms, blood counts, BM findings, and other laboratory parameters are usually sufficient to reveal the type and cause of a marked cytopenia. However, in patients with chronic mild cytopenia, it may be a challenge for the physician to establish a correct diagnosis. In such patients, laboratory features and findings often reflect a diagnostic interface, so that criteria that are otherwise robust may hardly be applicable or are not helpful. Even if the BM is examined, the diagnosis often remains uncertain in these patients. In addition, more than one potential cause of cytopenia may be present, especially in the elderly. A myelodysplastic syndrome (MDS) or another BM disorder, but also an overt autoimmune or other inflammatory disease, may develop during follow-up in these patients. A key problem is that in an early phase of MDS, most laboratory and clinical signs are “nonspecific.” One of the very few reliable peripheral blood parameters distinguishing between an early or “pre-phase” of MDS and most other causes of a mild cytopenia are the numbers of circulating colony-forming progenitor cells. In addition, flow cytometric and molecular investigations may sometimes assist in the delineation between clonal and reactive conditions underlying mild cytopenias. This review provides an overview of diagnostic approaches and algorithms for patients with mild unexplained cytopenia(s).
Introduction
Blood cell production is tightly controlled by several complex, interacting biological networks, including the growth factor and cytokine networks, the hematopoietic microenvironment, the immune system, other intercellular communication networks, and diverse intracellular communication (eg, signaling) networks.1–4 In addition, genetic, epigenetic, and metabolic factors contribute to the regulation of blood cell production.1–4 Finally, blood counts are also controlled by several negative-regulatory and clearance mechanisms, including “natural aging” of cells, growth-inhibitory cytokines, immune-mediated clearance of aged or damaged “end cells,” and cellular (intrinsic) clearance mechanisms such as apoptosis or anoikis.5–8 All of these mechanisms may act together to control and maintain blood cell counts at a remarkably constant “physiologic” range in healthy persons throughout their lifetimes. However, in various pathologic states, one or more of these regulatory systems are less active, suppressed, destroyed, or hyperactivated, and thereby may cause or contribute to the development of cytopenia(s).
In many patients, the cause of cytopenia is obvious and it is not difficult to establish the correct diagnosis after a first examination. Traditionally, cytopenias have been classified as deficiency related (ie, a nutritional or hormone deficiency), immune mediated, BM failure based, or idiopathic cytopenias. When certain criteria are fulfilled and other causes have been eliminated with certainty by all standard investigations, including a BM examination, the latter have been termed idiopathic cytopenias of undetermined (uncertain/unknown) significance (ICUS).9–11
In some patients with mild cytopenia, especially elderly patients, more than one cause of cytopenia may be present, which is a challenge in daily practice. Another challenge is chronic mild cytopenia in the context of certain comorbidities that may or may not contribute to the cytopenic state. In some of these patients, the follow-up may reveal an underlying pre-phase of a myelodysplastic syndrome (MDS) or another BM disorder10,11 or an occult autoimmune (or other inflammatory) disease. In other patients, a chronic infection, mild hemolysis, or an endogenous defect in erythropoietin (EPO) production with or without impaired excretory kidney function is found.10–13
In this review, the pathogenesis and diagnosis of several conditions that may underlie chronic mild cytopenia(s) are reviewed. In addition, thresholds defining the normal blood count are discussed. It is important to note that the diagnostic algorithm for patients with mild “unexplained” cytopenia requires a multistep and often multidisciplinary approach, and thus requires collaboration between practitioners, specialists, and specialized hematology centers in which the full “armamentarium” of modern hematology can be applied if necessary. A straightforward diagnostic approach preventing multiple referrals is usually the most effective procedure for saving time and costs.
Normal and cytopenic blood counts
The normal blood count is defined by normal values in all 3 major hematopoietic cell lineages (leukocytes, hemoglobin, and platelets) plus normal differential counts in the white cell compartments. A complete blood picture also includes the reticulocyte count, which should be normal (ie, a normal blood count) or appropriately compensated for the degree of anemia. Reticulocyte counts should be reported in absolute values or, if only reported as percentage values, should be translated into absolute counts. The term cytopenia refers to a condition in which one or more lineages show abnormally low counts. The threshold criteria discriminating normal, low, and diagnostic for various BM failure syndromes such as MDS vary (Table 1). In addition, threshold levels differ depending on age, sex, sea level, and race. Likewise, the neutrophil and leukocyte counts are higher in the white population compared with those of African descent. Therefore, “normal” reference ranges have to be established in each laboratory based on that laboratory's patient population. There is also an ongoing debate regarding normal WBC and neutrophil counts defined by the World Health Organization (WHO). In fact, several experts believe that it may be wise to revisit the threshold for neutropenia by reducing it from 1.7 × 109/L of blood to a lower count (eg, 1.5 × 109/L) to make sure that “normal” will include a majority of healthy subjects in various populations and countries. It is important that these thresholds are not identical to cutoff levels used as criteria to diagnose or grade MDS or other BM failure syndromes or to diagnose ICUS (Table 1).
Normal blood counts and definition of cytopenias

Hb indicates hemoglobin; ANC, absolute neutrophil count; PLT, platelet count; IPSS, International Prognostic Scoring System; and IPSS-R, revised IPSS.
*Diagnostic cytopenias refer to threshold levels used as criteria for various BM failure syndromes. In this table, thresholds used as criteria to diagnose MDS and ICUS are shown. It is important to note that apart from these lower blood count levels, other criteria have to be fulfilled as well to establish the diagnosis MDS or ICUS and the cytopenia must be persistent (> 6 months) to count as a criterion.
†The definition and criteria of ICUS were published by a consensus group in 2007.9
‡Based on the WHO classification, a MDS may also be diagnosed in patients with less severe cytopenia if morphologic and cytogenetic evidence clearly argue for MDS.
§The proposal of the IWGM-MDS group was presented at several MDS meetings.
¶In the white population, ANC values are higher than in people of African descent, in whom ANC levels are often < 1800/μL of blood in healthy controls.
Initial approach to the patient with chronic mild cytopenia
A detailed case history should exclude potential mutagenic events, toxin exposure, drug/alcohol intake, and known infectious diseases, including hepatitis, EBV, and HIV. Although most are acquired, some of the chronic mild cytopenias may be inherited disorders. The most frequent causes of chronic mild cytopenias in the Western world are hepatic disorders (eg, neutropenia and/or thrombocytopenia) and chronic inflammatory diseases (eg, anemia of chronic disease). A slightly impaired kidney function, as is often seen in the elderly, may be accompanied by chronic mild anemia. A summary of major causes of chronic mild cytopenia (lasting for at least 6 months) is provided in Table 2. It is essential to document the chronology and duration of cytopenias and to ask for potential causes in each case. Weight loss, fever, and other key symptoms may guide the physician in the initial evaluation.
Major causes of chronic mild cytopenia

NHL indicates non-Hodgkin lymphoma; AIHA, autoimmune hemolytic anemia; and AOE, anemia of the elderly.
*The normal neutrophil count of the people of African descent is lower than that in whites. Therefore, the ethnic background may explain a “lower” neutrophil count. However, this should not automatically be judged as neutropenia. The real inherited neutropenias are rare conditions.
†When infiltration is extensive or other cofactors such as inflammation occur, an indolent BM neoplasms may well lead to or contribute to BM failure with cytopenia.
A thorough physical examination is performed in each case. The presence or absence of palpable splenomegaly and/or lymphadenopathy should be documented. In many cases, an ultrasound investigation of the liver and spleen is recommended.
A complete blood count with microscopic differential count should be performed at first presentation. In addition, routine laboratory examinations should be performed, including a complete chemistry profile with electrolytes, serum creatinine, lactate dehydrogenase, hepatic enzymes, albumin, Igs (ie, IgG, IgA, and IgM), free light chains, ferritin, fibrinogen, and C-reactive protein. Depending on the type and duration of cytopenia, additional laboratory examinations may be performed. These include, among others, Ab screen tests, virus serology, a vitamin B12 and folate level, serum EPO level, and a serum tryptase level. An “early” flow cytometric screen of peripheral blood cells is often initiated in case of suspected paroxysmal nocturnal hemoglobinuria (PNH) to screen for GPI-linked Ag deficiency and if occult T- or natural killer cell large granular lymphocytic leukemia is suspected in neutropenic patients. In the latter, blood cells are also examined for the presence of a clonal TCR rearrangement, although, depending on the method applied, this approach may yield false-positive results.
A most sensitive screen parameter is the number of CFU progenitor cells.14–17 This assay, although not available in most hematology centers as a routine parameter, is of great value in the evaluation of patients with unclear mild cytopenias. Clinically, it is important to know that normal numbers of both granulocyte-macrophage CFU (CFU-GM) cells and erythroid burst-forming units (BFU-E) make the diagnosis of MDS very unlikely (Figure 1).14–17 The disadvantage of the CFU assay is that it needs great experience and precision and a dedicated team to generate reproducibly constant results for routine diagnostic purposes.

Numbers of CFU progenitor cells in patients with MDS and in controls. Numbers of circulating CFU-GM cells (left panel) and BFU-E cells (right panel) were determined in a methylcellulose assay. Peripheral blood mononuclear cells were plated on day 0 and cultured for 14 days in the presence of cytokines (IL-3, GM-CSF, and EPO). Colonies were counted under an inverted microscope. Results show the numbers of CFUs/BFUs/μL blood and represent the ranges (bars), 50 percentile values (boxes), and median values (horizontal lines within boxes) in each group of patients (10-15 patients per group). The grey area indicates the normal range of CFU-GM and BFU-E cells. All investigations were performed within (as part of) the routine examination of cytopenic patients at the Medical University of Vienna. AIHA indicates autoimmune hemolytic anemia; RA, refractory anemia; RARS, RA with ring sideroblasts; and RAEB, RA with excess blasts.
Numbers of CFU progenitor cells in patients with MDS and in controls. Numbers of circulating CFU-GM cells (left panel) and BFU-E cells (right panel) were determined in a methylcellulose assay. Peripheral blood mononuclear cells were plated on day 0 and cultured for 14 days in the presence of cytokines (IL-3, GM-CSF, and EPO). Colonies were counted under an inverted microscope. Results show the numbers of CFUs/BFUs/μL blood and represent the ranges (bars), 50 percentile values (boxes), and median values (horizontal lines within boxes) in each group of patients (10-15 patients per group). The grey area indicates the normal range of CFU-GM and BFU-E cells. All investigations were performed within (as part of) the routine examination of cytopenic patients at the Medical University of Vienna. AIHA indicates autoimmune hemolytic anemia; RA, refractory anemia; RARS, RA with ring sideroblasts; and RAEB, RA with excess blasts.
Depending on initial blood counts and serology test results, further investigations may be performed. The key diagnostic approach is to investigate the BM to clarify the cause and etiology of the cytopenia. At that time, it is helpful to know what major differential diagnoses have to be considered and what BM parameters are required to establish or exclude these diagnoses. However, in many cases, the situation is unclear and the only possible approach is to screen with a broad panel of parameters and markers. Two considerations are crucial in this situation: First, it is important to provide all important (available) clinical and hematological information (including blood counts, key findings, age, and suspected differential diagnoses) to the specialists in their respective laboratories (ie, pathology, cytogenetics, flow cytometry, and molecular) when sending BM material. In certain cases, it may even be preferable to contact the specialist directly by phone or e-mail to discuss the “unclear” situation. Second, it is important to store BM and peripheral blood cells (if possible, intact cells, RNA, and DNA) and serum and/or plasma for further cellular, molecular, and serologic investigations as backup material.
In all patients with chronic mild cytopenias, BM investigations should include a thorough cytological examination on a good-quality smear, histological and immunohistochemical analyses, chromosome analysis, and, if possible, flow cytometric studies according to available recommendations.9,18–29 In the event of a poor-quality aspirate smear, a touch imprint of the fresh biopsy should be performed to have adequate cytologic material available for evaluation. Smears should be examined by standard stains, including May-Grünwald-Giemsa or W-Giemsa and Prussian blue stain for iron.18–22,29 Immunohistochemical stains should detect CD34+ precursor/blast cells and cells in all major hematopoietic lineages, including B and T cells, granulocytes, macrophages, megakaryocytes, and mast cells.18–20,21 In addition, fibrosis should be excluded by reticulin or trichrome stains or a Gömöri silver stain. Chromosome analyses should include conventional karyotyping in a first step.9,23–25 If the result is normal but a certain diagnosis is likely or has to be excluded (eg, MDS) or the result is questionable, FISH of the lesions specific for the suspected neoplasm should be performed in a second step.9,25 Sometimes, a small clone with a characteristic cytogenetic lesion (eg, typical for MDS) is found by FISH. In such cases, it may be necessary to reinvestigate the BM after a certain time interval to control and compare the clone size.
Flow cytometry should be performed according to established guidelines to screen patients with suspected hematopoietic malignancies, including MDS.26–28 If no particular underlying disease is suspected, flow cytometry should be used to investigate the percentage and phenotype of CD34+ blast cells and should screen for phenotypic aberrancies in major BM lineages. Sometimes, flow cytometry may reveal a small clone that exhibits characteristic (eg, PNH-like or MDS-like) features. In these patients, a reinvestigation of the BM after some time may be considered depending on other results and on the course of the cytopenia.
In many conditions, immunohistochemistry and flow cytometry cover the same markers and may yield exactly the same results. The decision on whether one or both techniques should be applied depends (apart from availability and center experience) on the suspected disease/condition, the quality of the sample(s), and the cell types involved. In many situations, proper selection of markers and techniques, cell-sample storage for later use, and stepwise evaluations may be cost-effective approaches.
Molecular genetic studies are usually not performed in patients with mild chronic cytopenia unless there are signs of an occult underlying lymphoma (Ig or/and TCR rearrangement) or a certain inherited disorder. However, in the future, advanced sequencing studies30–33 may reveal/confirm that BM cells are monoclonal (somatic or in the germline) and may also reveal the lesion(s) that is likely to contribute to the cytopenia recorded. In several patients with mild cytopenia (not fulfilling MDS or ICUS criteria), a neoplastic clone is established and has replaced normal BM cells. When accompanied by signs of dysplasia, this condition has been termed idiopathic dysplasia of unknown (or uncertain) significance (IDUS).10,11 In such patients, as well as in those with ICUS, clonal lesions may be detectable by advanced molecular studies rather than by cytogenetics or FISH. Therefore, when standardized appropriately, advanced molecular approaches may become standard in patients with chronic mild cytopenia, ICUS, or IDUS.
ICUS
The term ICUS was created to describe an unexplained persistent cytopenia in patients in whom (minimal) diagnostic criteria for MDS are not fulfilled.9–11,34–36 It is important to state that the diagnosis of ICUS can only be established when a BM investigation was performed and all other causes of cytopenia have been ruled out. It is of particular importance that diagnostic dysplasia (ie, > 10% in one or more lineages) is excluded in the peripheral blood and in the BM in these patients.9–11 This means that no prominent population of Pseudo-Pelger neutrophils are found in the peripheral blood and no prominent dysplasia (≥ 10% of cells) is found in any of the 3 major lineages in the BM. If possible, dysplasia should also be excluded by flow cytometric studies.9,26–28
ICUS involves 1, 2, or all 3 major hematopoietic cell lineages. Most patients with ICUS have unilinear mild cytopenia, namely idiopathic anemia of undetermined significance (ICUS-A), idiopathic neutropenia of undetermined significance (ICUS-N), or idiopathic thrombocytopenia of undetermined significance (ICUS-T).11 Rarely, an idiopathic bi/pancytopenia of undetermined significance (ICUS-BI/PAN) is found. Similar to MDS, the cytopenia must be substantial and must be recorded for a time period of at least 6 months to count as a criterion of ICUS.9–11 Cutoff levels defined for the diagnosis of ICUS are shown in Table 1. A consensus group has proposed the following threshold levels for ICUS in 2007: hemoglobin, 11 g/dL; neutrophils, 1.0 × 109/L; and platelets, 100 × 109/L of blood.9 Similar values have been proposed by the International Working Group on Morphology of Myelodysplastic Syndrome (IWGM-MDS; Table 1).34,35 Several diagnostic investigations, including karyotyping, histology, and immunohistochemistry, have to be performed to establish the diagnosis of ICUS.9–11 Likewise, the presence of an MDS-specific karyotype37 will change the diagnosis from ICUS to MDS. In some patients with ICUS, MDS or another BM neoplasm may indeed develop in the follow-up period9–11,34–36,38 ; therefore, the follow-up of patients with ICUS should be the same as that in patients with low-risk MDS.
So far, little is known about cellular and molecular mechanisms underlying the evolution of ICUS to MDS. In some cases, clonal hematopoiesis is found by the human androgen receptor assay (HUMARA) and in a few patients smaller (subdiagnostic) MDS-like clones are detected by FISH or by flow cytometry.9–11,36,38,39 Currently, deep-sequencing studies and other studies are performed in research laboratories to delineate aberration profiles in ICUS patients and to detect primary lesions responsible for the manifestation and evolution of the disease. However, these advanced technologies are not yet available for broader routine investigations in these patients.
One important additional cofactor may be the cytokine network. First, ICUS may result from the effects of various negative regulators of hematopoietic cell growth (eg, TNF-alpha or TGF-beta) produced by activated immune cells in an occult (inflammatory or neoplastic) disease. Another example is growth factor deficiency. The production of EPO in response to anemia may be impaired in elderly patients even if the secretory kidney function is normal.12,13,40 Such abnormal endocrine kidney function, resulting in low EPO levels, may contribute to the development of anemia and can indeed be found in (elderly) patients with ICUS.10,11,40 These patients are often diagnosed as having anemia of the elderly (AOE).12,13 In other words, AOE can be regarded as a subvariant of ICUS and, in many such cases, an inadequately low level of endogenous EPO (relative to the anemia) is found. In these patients, therapy with recombinant EPO may work in the same way as in patients with low-risk MDS with altered EPO production (low endogenous EPO levels).40
Discrimination between ICUS and low-risk MDS
An important question is how to discriminate between ICUS and low-risk MDS with certainty. First, it is important to apply all available criteria and parameters in these patients.9–11 Likewise, the use of FISH in patients with unexplained chronic cytopenia may indeed reveal a small clone. In these patients, clonal expansion (over time) may be documented by FISH in subsequent samples, so that the diagnosis changes from ICUS to MDS.9–11,36 If no karyotypic abnormality and no dysplasia is detected, the situation is more difficult. For these patients, it has been recommended to look for additional signs of BM failure, aberrant phenotypes, and other signs of a clonal disease process.
A reliable parameter for an impaired BM function is a reduced number of CFU progenitor cells (Figure 1). However, reduced numbers of CFU-GM and BFU-E cells are also found in patients with aplastic anemia, acute leukemias, and after a massive toxic event (eg, chemotherapy). With regard to clonality of BM cells, flow cytometry is a reliable parameter with high sensitivity. In fact, certain abnormalities in expression of cell surface (ie, leukocyte-differentiation) Ags determined by flow cytometry may suggest the diagnosis MDS in a cytopenic patient.26,27 These include an abnormal intensity or lack of expression of CD34, CD45, CD13, CD33, CD117, or HLA-DR; abnormal expression of CD11b or CD15; and/or the aberrant expression of CD5, CD7, or CD56 on myeloid progenitors.26,27 In addition, the loss of certain maturation markers on mature myeloid cells (eg, CD10 on neutrophils) or aberrant coexpression of immature myeloid markers such as CD34 may be indicative for a clonal disease process. In addition, abnormal side-scatter patterns, an increase in CD34+ cells, and a relative decrease in B-lymphoid progenitors has been described.26,27 However, abnormal flow patterns are also found in acute myeloid leukemia and even in myeloproliferative neoplasms. In addition, the BM undergoes natural aging that may also be accompanied by phenotypic changes and a shift toward myeloid (progenitor) cells.41
Screening for molecular lesions may also be a reliable approach to use to look for signs of clonal hematopoiesis in patients with ICUS. However, so far little is known about the type of lesions found in ICUS and about the diagnostic impact of these markers. Another approach is to apply the HUMARA assay.38 However, this assay is only applicable to female patients and is also positive in other clonal states. An emerging approach is to screen for molecular lesions by exome sequencing and other “omics-based” techniques.30,31 However, these approaches are expensive and not applicable in daily practice. In addition, little is known about specificity and sensitivity of these assays. Conversely, it is clear that the molecular heterogeneity and complexity of the progenitor cell pool in MDS and in the MDS-preceding type of ICUS and IDUS will require an advanced molecular approach in the future, and advanced sequencing and omics methods may be an optimal tool to use to address this important issue.
Discrimination between MDS and other causes of cytopenia
There are several causes that have to be considered in patients with chronic mild cytopenias, such as chronic inflammatory reactions, autoimmune diseases, infectious disorders, viral disorders, chronic hepatic or renal disorders, or chronic immune-mediated cytopenias.42,43 In addition, mild iron deficiency, vitamin B12 or folate deficiency, copper deficiency, and other deficiency syndromes may sometimes produce mild cytopenias, at least for a certain time (before they become more severe). A summary of common causes of mild chronic cytopenias is shown in Table 2. Depending on the blood count, differential counts, erythrocyte morphology, and erythrocyte indices, several laboratory parameters are applied to define the exact nature of the cytopenias and the underlying condition. Sometimes these parameters need to be interpreted with caution, especially in “comorbid” patients (eg, ferritin levels in patients with iron deficiency plus inflammation). In addition, the combination of certain parameters (rather than a single parameter or abnormality) is often indicative of an underlying condition (eg, MDS) and can thus be used to rule out or to establish certain causes of cytopenia. An example is macrocytosis, which may or may not be indicative of MDS depending on additional parameters such as lactate dehydrogenase and the reticulocyte count. A summary of parameters and parameter combinations from routine and specialized hematology laboratories that suggest certain causes of cytopenia is shown in Table 3.
Parameters and parameter combinations useful for the discrimination between MDS and other conditions producing anemia
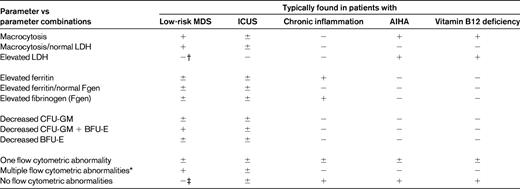
LDH indicates lactate dehydrogenase.
*Aberrant flow patterns suggesting the presence of a MDS are described in the available literature.26–28
†An elevated LDH in low-risk MDS is rarely found. In these patients, LDH levels result from a comorbidity, a small PNH clone, G-CSF therapy, or clonal cell expansion with imminent progression to high-risk MDS.
‡In rare patients with MDS, no flow cytometric abnormalities are found.
Multiple causes and conditions that act together to cause cytopenia and produce overt MDS
In many patients, more than one cause (ie, pathologic condition) may contribute to the development of a cytopenia. This holds true especially for patients with comorbidities, such as elderly patients. Even in the absence of a known comorbidity, elderly patients may have an underlying occult condition predisposing for the development of cytopenia(s). One example is the low EPO production (despite otherwise apparently normal kidney function) in elderly patients that may contribute to the development of anemia in, and thus manifestation of, MDS.12,13,44 In the presence of other cofactors such as clonal hematopoiesis, such predisposing conditions will lead to the development of an overt cytopenia or act together to promote cytopenia.
Recent data suggest that BM cell dysplasia alone may not necessarily lead to cytopenias in elderly patients, even if normal BM cells are completely replaced by the dysplastic clone.10,11,44 This situation has been termed ideopathic dysplasia of unknown significance (IDUS) because the outcome remains uncertain. Some of these patients develop only mild cytopenias, not fulfilling the criteria of a MDS. Other patients with IDUS develop marked cytopenia and thus MDS after a variable latency period (estimated range, months to decades). Several different mechanisms may underlie the manifestation of MDS in a patient with IDUS. One is malignant transformation of cells that no longer respond to regulatory cytokines such as EPO. Another cause are comorbidities such as renal anemia or a hepatic disease. Finally, a low production of EPO alone may be sufficient to cause manifestation of MDS in a patient with IDUS.44 In other words, the status “IDUS” depends on an otherwise healthy tissue environment and normal production of regulatory cytokines such as EPO, as well as on an appropriate response of (clonal) erythropoietic progenitor cells to EPO.10,11,44 Alternatively, in some patients with IDUS, residual normal erythropoietic progenitors may respond to EPO. If, in patients with IDUS, EPO production decreases because of aged kidneys or overt renal failure (demonstrable by low endogenous EPO), the subdiagnostic lesion IDUS will manifest as an overt MDS.44 In other words, the relatively ineffective (clonal) BM and the renal anemia (or AOE) act together as responsible lesions for the manifestation of MDS. A clinical proof of this concept is that endogenous EPO levels are low in many cases (especially in the elderly) and that treatment with exogenous EPO improves or even corrects hemoglobin levels in many MDS patients who have relatively low endogenous EPO levels.45,46 However, in addition to a low EPO level, there are other cofactors that may contribute to the development of cytopenia and thus manifestation of MDS in elderly patients with IDUS. Among these are chronic inflammatory disorders, hemolysis (eg, in the context of a PNH clone), hormonal disorders, and hepatic disorders. In all these comorbidity states, when present in the absence of MDS, the levels of circulating progenitor cells (CFU-GM and BFU-E) are usually normal, whereas progenitor counts are abnormally low in most patients with MDS or IDUS, which is of diagnostic importance. Therefore, the CFU assay, although not used in most centers on a routine basis, should be considered as an important pre-invasive (functional) screen parameter in patients with unexplained mild cytopenia.
Summary and future perspectives
Chronic unexplained mild cytopenias are a challenge in clinical practice. In many cases, no underlying cause is detected even after extensive evaluations, including BM investigations. In these patients, the diagnosis of ICUS is appropriate. In other patients, an underlying nonhematologic or hematologic disease, often in the form of an MDS, is detected after extensive investigations or in the follow-up period. Especially in elderly patients, an overt MDS may develop after a variable latency period. Although no parameter is specific for any cause of cytopenia in these patients, the numbers of CFU progenitor cells and flow cytometric studies may yield additional information useful for the delineation of MDS and other BM disorders from nonhematologic causes of cytopenia.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Peter Valent, MD, Department of Internal Medicine I, Division of Hematology & Hemostaseology and Ludwig Boltzmann Cluster Oncology, Medical University of Vienna, Waehringer Guertel 18-20, A-1090 Vienna, Austria; Phone; 43-1-40400-5488; Fax: 43-1-40400-4030; e–mail: peter.valent@meduniwien.ac.at.
