
Abstract
Monoclonal gammopathy of undetermined significance (MGUS) is an asymptomatic plasma cell disorder occurring in 4.2% of adults > 50 years of age, which can progress into symptomatic diseases either through proliferation of the plasma cell clone, giving rise to multiple myeloma and other lymphoplasmacellular neoplasms, or through organ damage caused by the monoclonal protein, as seen in light-chain amyloidosis and related conditions. Differential diagnosis of asymptomatic and symptomatic monoclonal gammopathies is the determinant for starting therapy. The criteria for determining end-organ damage should include markers of organ injury caused by the monoclonal protein. Patient assessment and optimal follow-up are now performed using risk stratification models that should also take into account the risk of developing AL amyloidosis. Patients with low-risk MGUS (approximately 40% of all MGUS patients) need limited assessment and very infrequent follow-up. The ongoing development of novel molecular biomarkers and advanced imaging techniques will improve the identification of high-risk patients who may benefit from early therapeutic intervention through innovative clinical trials.
Introduction
Robert Kyle coined the term monoclonal gammopathy of undetermined significance (MGUS) in 1978 after the observation that asymptomatic patients with a monoclonal protein (M-protein) had higher risk of developing multiple myeloma (MM), Waldenström macroglobulinemia (WM), light-chain amyloidosis (AL), or related conditions. MGUS is a premalignant clonal disorder that is present in more than 4% of the general white population older than 50 years of age1,2 and is associated with a 1%/y risk of progression to MM or related malignancies. Because the transition from MGUS into a malignant, symptomatic condition is an evolving process,3 the differential diagnosis between MGUS and these diseases is frequently challenging. MGUS may progress toward symptomatic conditions, requiring the initiation of treatment, through 2 general mechanisms (Figure 1): (1) progression of the proliferative characteristics toward smoldering MM (SMM), MM, and other lymphoplasmacellular disorders, which account for approximately 90% of the progression; or (2) the development of end-organ damage caused by the M-protein such as AL amyloidosis, light chain deposition disease (LCDD), and other rarer conditions, which account for approximately 10% of the progression. Because most MGUS progression occurs toward MM, little attention has been dedicated to the development of other disorders caused by the M-protein.

Conditions associated with an M-protein. Although MGUS is a premalignant condition, approximately 40% of all MGUS patients are considered low-risk MGUS and have a lifetime risk of progression of only 2%.32 Acquisition of somatic genetic abnormalities in the tumor cells and changes in the BM microenvironment may lead to progression to SMM and MM. SMM includes patients with premalignancy and patients with early asymptomatic malignancy. The clone may produce a protein with altered conformation, which may aggregate improperly, causing progressive organ dysfunction. These conditions include AL amyloidosis, LCDD, and type I cryoglobulinemia. Other conditions, such as type II cryoglobulinemia, chronic cold agglutinin disease, and autoimmune neuropathies, are caused by the autoantibody activity of the M-protein, which in most cases is an IgM. Finally, other rare diseases are associated with monoclonal gammopathies, but their pathogenesis is still unclear. This is the case for POEMS syndrome, scleromyxedema, and Schnitzler syndrome.
Conditions associated with an M-protein. Although MGUS is a premalignant condition, approximately 40% of all MGUS patients are considered low-risk MGUS and have a lifetime risk of progression of only 2%.32 Acquisition of somatic genetic abnormalities in the tumor cells and changes in the BM microenvironment may lead to progression to SMM and MM. SMM includes patients with premalignancy and patients with early asymptomatic malignancy. The clone may produce a protein with altered conformation, which may aggregate improperly, causing progressive organ dysfunction. These conditions include AL amyloidosis, LCDD, and type I cryoglobulinemia. Other conditions, such as type II cryoglobulinemia, chronic cold agglutinin disease, and autoimmune neuropathies, are caused by the autoantibody activity of the M-protein, which in most cases is an IgM. Finally, other rare diseases are associated with monoclonal gammopathies, but their pathogenesis is still unclear. This is the case for POEMS syndrome, scleromyxedema, and Schnitzler syndrome.
Recognition of an M-protein
Most M-proteins are detected incidentally during the investigation of other conditions. There is consensus that population-based screening should not be performed considering the generally low risk of progression to a malignant disorder and the related emotional burden. However, in the presence of clinical suspicion of MM, WM, AL amyloidosis, or related disorders, the clinician should always screen for an M-protein. Although virtually 100% of patients with MM and WM can be identified by serum electrophoresis and quantitative serum free light chain (FLC) assays, serum and urine immunofixation are also required to achieve the best diagnostic sensitivity for other serious conditions such as AL amyloidosis or LCDD.4,5
Prevalence and risk factors
Using the serum FLC assay and immunofixation electrophoresis, Mayo Clinic investigators determined the overall age- and sex-adjusted prevalence of conventional MGUS in persons 50 years of age or older in Olmsted County to be 3.4%.1,6 Considering that the prevalence of light-chain MGUS is 0.8%, the overall MGUS prevalence is 4.2% (95% confidence interval, 3.9-4.5).6 Studies in both white and Japanese populations demonstrate a clear increase in prevalence with age. The prevalence is also affected by sex: 3.7% and 2.9% in white men and women, respectively, and 2.8% and 1.6% in Japanese men and women, respectively. MGUS is twice as prevalent in black adults (5.9%-8.4%).7
The etiology of MGUS is unknown, and previous studies support a role of both genetic and environmental factors. A family history of MGUS or MM increases the risk of MGUS by approximately 3.3- and 2-fold, respectively, suggesting a shared environmental and/or genetic effect.8–10 Radiation exposure, particularly at a younger age, increases the risk of MGUS significantly, but not that of malignant progression.11 The use of certain pesticides,12 as well as African ethnicity, obesity, and increasing age are associated with an excess risk of MGUS.13
Distinct clinical subtypes of MGUS and related disorders
Large epidemiological and clinical studies have led Mayo Clinic investigators to define 3 distinct clinical subtypes of MGUS, each with a different mode of progression: non-IgM MGUS, by far the most common; IgM-MGUS; and light-chain MGUS (Table 1).14 Non-IgM MGUS and light-chain MGUS have a plasma cell (PC) phenotype and can progress to MM or related PC disorders, whereas the 15%-20% of patients with IgM-MGUS usually have a lymphoid or lymphoplasmacytoid phenotype and can progress to WM, lymphoma, or other neoplastic lymphoproliferative disorders. IgM-MGUS patients have a distinct molecular pathogenesis compared with PC MGUS (see Ghobrial15 ). The best-characterized subtype is non-IgM MGUS, in which the M-component isotype can be IgG (69%), IgA (11%), or biclonal (3%); IgD and IgE are rare.1 All types can evolve into MM, AL amyloidosis, and related disorders, with a rate of approximately 1%/y. Light-chain MGUS is a recently identified asymptomatic condition characterized by an abnormal FLC ratio (κ:λ) of < 0.26 or > 1.65 and an increased concentration of the involved light chain.6 Although this entity is now being recognized more frequently, as reported by German investigators,16 its natural history requires further studies.
Disease definitions for the non-IgM monoclonal gammopathies
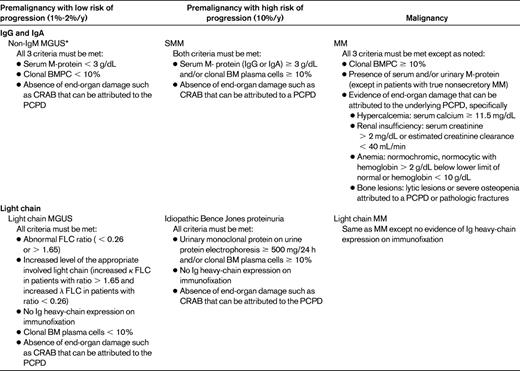
PCPD indicates PC proliferative disorder.
*Occasionally, patients with IgD and IgE monoclonal gammopathies have been described and are also considered part of this category.
Adapted with permission from Rajkumar et al.14
Biological progression from MGUS to MM
MGUS is a premalignant clonal disorder that in virtually all patients precedes the development of MM and related disorders, as documented by 2 independent studies.17,18 Multistep genetic and microenvironmental changes lead to the transformation of these clonal cells into a malignant neoplasm,19,20 a detailed description of which is beyond the scope of this review. Briefly, the primary cytogenetic abnormalities can be divided into 2 partially overlapping, general entities according to the chromosome number: (1) hyperdiploidy in approximately 50% of cases, with trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21, and (2) nonhyperdiploid (in the remaining 50%), which are often associated with a translocation event at the IgH locus (on chromosome 14) with recurrent chromosomal patterns and oncogene deregulation involving Cyclin D, MAF, and MMSET/FGFR3. Dysregulation of a Cyclin D gene seems to be a unifying event in virtually all MGUS and MM clones.19 Progression of MGUS to MM is probably secondary to a random, unknown, second “genetic hit” involving complex karyotypic abnormalities. Two events, deletion chr13 and activating K-RAS mutations, may be associated with the transition from MGUS to MM in some patients, whereas increased MYC expression and sometimes MYC locus rearrangements may be involved more commonly in this transition.21,22 Because these changes occur at different stages, they may serve as potential markers of disease progression. Epigenetic dysregulation, such as alterations in miRNA expression and gene methylation modifications, have also been reported.21 In addition to genetic abnormalities of PCs, abnormal interactions with the bone microenvironment result in bone lesions and aberrant angiogenesis associated with disease progression.
Risk stratification
At present, there are no reliable biologic markers that predict which individual with MGUS will progress to MM or related conditions. In the absence of such markers, MGUS is currently risk stratified based on clinical variables identified through epidemiological studies. Two predictive risk models for MGUS to MM have been developed by the Mayo Clinic and the Spanish study group investigators (Figure 2). The Mayo Clinic model is centered on serum protein abnormalities and identifies 3 major risk factors for progression: non-IgG isotype, serum M-component concentration > 1.5 g/dL, and an abnormal FLC ratio.23 The stratification of 1148 patients was: 39% low risk (no factor abnormal), 36.5% low-intermediate risk (any 1 factor abnormal), 20% high-intermediate risk (any 2 factors abnormal), and 4.5% high-risk (all 3 factors abnormal).23 The Spanish study group model is based on multiparametric flow cytometry techniques to identify aberrant PC (aPC) populations.24 An aPC/normal BMPC ratio of > 95% is associated with a higher risk of progression. MGUS risk factors for progression are aPC/BMPC > 95% and DNA aneuploidy. Using these 2 criteria, the stratification was: 46% score 0, 48% score 1, and 6% score 2.24 Furthermore, Bladé et al reported that approximately 10% of patients with MGUS show a progressive increase in M-protein leading to SMM and finally to symptomatic myeloma.25 The “evolving” MGUS would be consistent with “slowly evolving” or “early” myeloma from the time of its appearance, in contrast to the stable or “true” MGUS. The evolving subtype of MGUS has an independent prognostic value from having > 95% aPC seen on flow cytometry; nevertheless, the latter has a stronger prognostic power and the advantage that it can be used upfront.26 No risk stratification has been developed for light-chain MGUS; however, the progression rate of this condition is similar to that of Mayo Clinic low-risk MGUS.6

Risk-stratification schemes for MGUS according to the 2 major models. In the Mayo Clinic model, the following features are considered to be adverse risk factors: M-protein concentration ≥ 1.5 g/dL, non-IgG isotype, and an abnormal serum FLC ratio (normal reference, 0.26-1.65). Patients with 0, 1, 2, or 3 risk factors are considered low-, low-intermediate, high-intermediate, and high-risk, respectively. The Spanish model uses multiparametric flow cytometry of BM aspirates to differentiate aberrant from normal PCs. PCs characteristically express CD138 and intense (bright) CD38. The features of aPCs include decreased CD38 expression, expression of CD56, and the absence of CD19 and/or CD45. Risk factors for progression are ≥ 95% aPCs/BMPC and DNA aneuploidy.
Risk-stratification schemes for MGUS according to the 2 major models. In the Mayo Clinic model, the following features are considered to be adverse risk factors: M-protein concentration ≥ 1.5 g/dL, non-IgG isotype, and an abnormal serum FLC ratio (normal reference, 0.26-1.65). Patients with 0, 1, 2, or 3 risk factors are considered low-, low-intermediate, high-intermediate, and high-risk, respectively. The Spanish model uses multiparametric flow cytometry of BM aspirates to differentiate aberrant from normal PCs. PCs characteristically express CD138 and intense (bright) CD38. The features of aPCs include decreased CD38 expression, expression of CD56, and the absence of CD19 and/or CD45. Risk factors for progression are ≥ 95% aPCs/BMPC and DNA aneuploidy.
The actual risk of developing M-protein–related diseases and in particular AL amyloidosis, the most prevalent of these conditions, is poorly defined. Available data were obtained in patients with MGUS not including light-chain MGUS. In these cohorts of patients with intact Igs, it is known that evolution to AL amyloidosis accounts for approximately 10% of all progressions.3,27 However, 42% of our 1399 patients with AL amyloidosis had a light-chain–only M-protein; in the Mayo Clinic population, light-chain–only M-protein was present in almost 40% of patients.28 It seems unlikely that these cases originate from light-chain MGUS, considering the very low rate of progression to MM 0.3%/y (no patient developed AL amyloidosis) in the original series.6 In the recently reported German light-chain MGUS population, there was no progression during 5 years of follow-up.16
Differential diagnosis
The discrimination between asymptomatic and symptomatic conditions due to end-organ damage is the current determinant for starting therapy. Basically, it is end-organ damage that distinguishes MGUS from malignant conditions, so our discriminatory capability depends upon the quality of the tools available to detect end-organ damage.
Differentiating MGUS from SMM and MM
According to the diagnostic criteria for the 3 conditions (Table 1), the differential diagnosis is based on M-protein concentration, BMPC percentage, and end-organ damage defined by the hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) criteria. The reduction of uninvolved Igs in serum, the presence of Bence Jones proteinuria, or FISH abnormalities are of little help in distinguishing between patients with MGUS and those with MM, because these abnormalities may be present in both conditions. Metaphase cytogenetic studies rarely reveal an abnormal karyotype in MGUS because of the low proliferative rate and the small number of PCs. Symptomatic MM is often associated with circulating monoclonal PCs in the peripheral blood.29
According to the 2010 International Myeloma Working Group (IMWG) guidelines,2 MGUS is defined as having serum M-protein < 3 g/dL, clonal PC population in BM < 10%, and absence of end-organ damage according to the CRAB criteria (Table 1).2 However, because MGUS is a condition of the elderly, concomitant diseases can confound the distinction.
Hypercalcemia.
Hypercalcemia may be caused by hyperparathyroidism that should be considered in the absence of skeletal lesions and moderate, stable, hypercalcemia and should be confirmed by the measurement of serum parathyroid hormone.
Renal disease.
Renal disease and renal damage may be sustained by diabetes and hypertension. Because mild and moderate kidney injury is poorly assessed from serum creatinine alone, it is recommended to routinely measure the estimated glomerular filtration rate (eGFR), which is calculated from serum creatinine using the Modification of Diet in Renal Disease (MDRD) study equation.30 In general, eGFR is more sensitive at detecting kidney injury than a creatinine cutoff > 2.0 mg/dL and eGFR use should be encouraged among hematologists. If renal involvement is the sole criterion for starting therapy, with eGFR < 40 mL/min and minimal albuminuria, a renal biopsy is indicated to distinguish between light-chain–related kidney damage and renal damage unrelated to hematologic diseases. In the presence of high urinary albumin, AL amyloidosis and monoclonal Ig deposition disease should be considered and appropriate histology assessment should be pursued.31 Patients with renal involvement should be followed in collaboration with the nephrologist.
Anemia.
Anemia may be multifactorial in the elderly due to iron, folic acid, and vitamin B12 deficiency; nutritional imbalance; occult gastrointestinal bleeding; anemia of chronic disease; or, less frequently, myelodysplastic syndrome.
Bone lesions.
Bone lesions require scrutiny because diffuse osteoporosis is common in the elderly, particularly in women, and may lead to compression fractures. In these cases, thorough evaluation with an MRI is required, if a focal myelomatous lesion is detected, then the patient has symptomatic myeloma requiring treatment. If the fracture is the result of osteoporosis, then other criteria should be considered to diagnose symptomatic myeloma.32 Isolated, asymptomatic lytic bone lesions may be caused by a bone cyst or angioma. In addition, metastatic carcinoma associated with MGUS should be excluded. Based on current guidelines, a skeletal survey, including chest, skull, humeri, femora, pelvis, and anteroposterior and lateral images of the whole spine, remains the standard imaging modality to detect osteolytic lesions in MGUS (also considering its wide availability at modest cost).32 However, this technique is rather insensitive for the detection of osteolytic lesions because it requires at least 30% cortical bone destruction. Whole-body MRI and whole-body PET/CT using fluorodeoxyglucose (18F-FDG) are promising in distinguishing patients with active myeloma from those with MGUS or SMM, but their use must be better defined by further studies.33,34 Several bone metabolism markers are correlated with skeletal involvement and disease progression; however, further research is needed to assess their utility for the early detection of bone disease.
Studies using gene-expression profiling and miRNA expression to distinguish MGUS from MM are ongoing. However, more data are needed before these markers can be used in the clinic.20,21
An intermediate stage of disease between asymptomatic MGUS and MM is SMM, which is characterized by higher cutoff M-protein and BM PC values while maintaining the lack of end-organ damage (Table 1).2 From a biological point of view, SMM is a heterogeneous entity that includes patients with stable MGUS-like conditions and patients with early asymptomatic MM. It was reported recently that in 95% of patients with SMM presenting with 60% or greater BM PCs, the clinical course is characterized by progression to symptomatic MM within 2 years. These patients should be considered to have myeloma that requires therapy at the time of diagnosis.35 The risk of progression from SMM to MM is 10%/y for the first 5 years, 3% for the next 5 years, and 1% for the subsequent 10 years.36 This is in contrast to MGUS patients, in whom the risk of transformation remains constant over time.3 Risk-stratification criteria for progression from SMM to MM have been developed by the Mayo Clinic investigators36,37 and by a Spanish study group.24 The risk factors for progression of SMM include M-protein size, percentage of BMPCs, and an abnormal FLC ratio in the Mayo Clinic guidelines and the proportion of phenotypically abnormal BMPCs and immunoparesis in the Spanish guidelines. In addition, evolution pattern (evolving vs nonevolving) and MRI abnormalities are predictors of progression. Increasing anemia is the most reliable indicator of progression.38
Differentiating MGUS from M-protein–related diseases
Patients with MGUS can develop severe end-organ damage caused by structural characteristics of the M-protein or may be related to poorly understood paraneoplastic syndromes that are independent of the size and proliferation of the PC clone. These disorders, caused by “dangerous small B-cell clones,”39 can develop silently during follow-up, are often overlooked by clinicians, and may become manifest when organ function is already irreparably compromised. In this review, we focus on AL amyloidosis and light-chain deposition disease. Other disorders include conditions related to antibody activity of the M-protein, cryoglobulinemia, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome, acquired Fanconi syndrome, Schnitzler syndrome, and scleromyxedema (see Gertz and Buadi40 ). The clinical presentation of these diseases and clues for early diagnosis are summarized in Table 2.
M-protein–related disorders other than AL amyloidosis

Rarer conditions with related pathogenesis and overlapping clinical presentation are reported in parentheses.
CRP indicates C-reactive protein; MCV, mean corpuscular volume; and SAA, serum amyloid A protein.
In AL amyloidosis, an abnormal monoclonal light chain causes systemic proteotoxicity by misfolding, aggregating in fibrils, and interacting with and depositing in tissues.41 Unlike most other PC dyscrasias, the λ isotype accounts for 75% of the amyloidogenic light chains and 3 Vλ genes, IGLV2-14 (λII), IGVL6-57 (λVI), and IGLV3-1 (λIII) contribute to encoding almost 60% of amyloidogenic λ light chains.42–44 The clinical presentation depends on organ involvement and is described in Table 3. LCDD, which was the subject of a recent review,45 is characterized by nonfibrillar, Congo red–negative deposition of monoclonal light chains. In more than 90% of cases, κ light chains, most often of the VκIV subgroup, are responsible for LCDD.46 The kidney is the target organ, with proteinuria, microscopic hematuria, hypertension, and often rapidly declining renal function. In contrast to light-chain cast nephropathy, the degree of renal failure is not related to the level of Bence Jones proteinuria.
Clinical presentation of AL amyloidosis
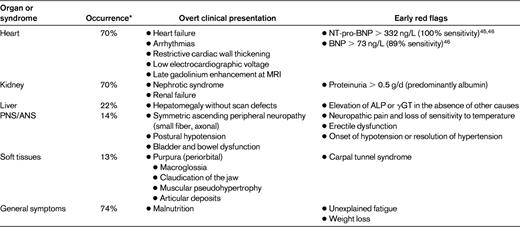
ALP indicates alkaline phosphatase; ANS, autonomic nervous system; γGT, γ-glutamyl transpeptidase; and PNS, peripheral nervous system.
*Data from 1399 patients diagnosed at the Pavia Amyloidosis Research and Treatment Center.
The differential diagnosis of M-protein–related diseases is based on early recognition of organ damage. Therefore, early diagnosis requires monitoring markers of initial organ dysfunction in patients at risk (Tables 2 and 3). This is particularly important in cardiac amyloidosis, because when symptoms become evident, irreversible cardiac damage has often already occurred. Conversely, the N-terminal fragment of pronatriuretic peptide type B (NT-pro-BNP) is a very sensitive47,48 marker of cardiac dysfunction in AL amyloidosis, and early increases precede by several months the onset of symptoms and the classical echographic features of heart involvement.49 However, natriuretic peptides are not specific markers of amyloid heart involvement; they can also be elevated in other cardiac diseases, particularly atrial fibrillation, and in renal failure. The concentration of BNP is less influenced by renal failure, but BNP is less sensitive than NT-pro-BNP in detecting cardiac amyloidosis.48 Detecting renal damage requires monitoring for albuminuria. In LCDD, albuminuria is often accompanied by hematuria and declining renal function.
The diagnosis of amyloidosis requires a tissue biopsy to demonstrate the presence of deposits with green birefringence under polarized light when stained with Congo red. Organ biopsy is seldom required and the diagnosis can usually be established by less invasive biopsies, such as those of the abdominal fat, minor salivary glands, BM, or rectum. It cannot be overemphasized that the demonstration of amyloid deposits in a patient with a monoclonal gammopathy is not conclusive evidence of AL amyloidosis. The concomitant occurrence of MGUS and non-AL-type amyloidosis (eg, hereditary, reactive to chronic inflammation, or senile amyloidosis), requiring completely different treatments, is possible and the demonstration of the light-chain origin of amyloid deposits in tissues is mandatory. This can be achieved in referral centers by direct proteomic characterization of the amyloid deposits50,51 or by immunoelectron microscopy. Light-microscopy immunohistochemistry, although useful when performed by highly specialized pathologists,52 is generally less accurate.
LCDD is usually diagnosed by renal biopsy showing nodular sclerosing glomerulopathy by light microscopy, diffuse linear staining of glomerular and tubular basement membranes for κ or λ light chains by immunofluorescence, and nonfibrillar electron-dense deposits by electron microscopy.45 Deposits must be Congo red–negative and stain for a single light chain.
Management of patients with MGUS
The present approach to a patient with clearly defined MGUS is a prudent “watch and wait” strategy that monitors patients on the basis of their risk category.2 The watch and wait approach is based on 2 main considerations: (1) negative results of clinical trials in which early treatment interventions using standard chemotherapy or strategies focused on bone remodeling were not associated with survival benefit,20,38 and (2) the low rate of progression in low-risk MGUS (only 2% at 20 years) and low-risk SMM (4%-25% at 5 years).24,37
Standard investigative workup
Although consensus statements and guidelines for the management of MGUS have been described previously,53,54 for the first time, the 2010 IMWG guidelines propose differential assessment and follow-up on the basis of the Mayo risk categories.2 In patients in whom an M-protein has been identified through serum electrophoresis and immunofixation, a few standard investigations are recommended, with additional studies graduated according to the risk.
A complete history and physical examination focusing on symptoms and findings that might suggest MM or AL amyloidosis should be performed. A complete blood count, serum calcium and creatinine (with eGFR) values, and a qualitative test for urine protein are also required. In the presence of proteinuria, electrophoresis and immunofixation are indicated.2 Serum FLC and κ/λ ratio should be determined for risk assessment (Figure 2).
In subjects with low-risk MGUS (approximately 40% of all MGUS patients), baseline BM examination and skeletal radiography are not routinely indicated if clinical evaluation, complete blood count, and serum creatinine and calcium values suggest MGUS. Patients should be followed with serum protein electrophoresis in 6 months and, if stable, every 2-3 years or when symptoms suggestive of a PC malignancy arise. Furthermore, risk-stratification studies of MGUS using the Mayo Clinic scheme suggest that routine annual follow-up of MGUS may not be required in low-risk MGUS patients.55 We usually discharge these patients back to family physicians.
In subjects with intermediate- and high-risk MGUS, a BM aspirate and biopsy should be carried out at baseline, along with metaphase cytogenetics, FISH, and bone-imaging studies such as skeletal survey. If available, a PC-labeling index and a search for circulating PCs using flow cytometry are useful. If the results of these tests are satisfactory, patients should be followed with serum protein electrophoresis and a complete blood count in 6 months and then annually for life. There is consensus that in high-risk patients, the decision to treat should be based on CRAB criteria. Patients with clearly defined MGUS do not need initiation of therapy regardless of any associated risk factors, including cytogenetics abnormalities, except on specifically targeted protocols.56
In AL amyloidosis, the progression of organ damage may be completely silent until the late stage. Amyloid cardiomyopathy can develop and progress during monitoring, even in the hands of an expert, because currently recommended tests cannot detect heart involvement. The majority (83% in our series) of AL patients have abnormal FLC studies. Therefore, patients with MGUS and an abnormal FLC ratio should be monitored to detect organ damage caused by the light chain, particularly heart involvement, which is the main prognostic determinant of survival, and renal damage. This can be performed efficiently by measuring NT-pro-BNP and urine albumin at MGUS presentation and at each follow-up visit. If these tests are positive or if there is the presence of any other red flags (Table 3), a procedure to diagnose AL amyloid should be pursued. Amyloid deposits can be found in the BM of patients with MGUS in the absence of organ damage. These subjects with “asymptomatic amyloidosis” do not require treatment, but should be carefully followed to detect the possible onset of organ dysfunction in a timely manner.
At each subsequent visit, testing should include serum protein electrophoresis, 24-hour urine protein excretion with urine electrophoresis, hemoglobin level, serum creatinine (eGFR), and calcium concentration.2 In patients with an abnormal FLC ratio, the NT-pro-BNP level should also be determined. The M-protein usually remains stable until myeloma develops. Patients with evolving MGUS but with M-protein size still in the MGUS range can be followed with annual visits; however, when they fulfill the SMM criteria (M-protein size ≥ 3 g/dL), follow-up should be that normally used in SMM: in general, every 4-6 months at the clinician's discretion.
The 2010 IMWG guidelines also report optimal management of patients with SMM and suggest that preventive clinical trials need to be considered for patients with high-risk SMM.2
Conclusions and perspectives
There is a need for more sensitive biomarkers to detect the malignant condition earlier, with the aims of: (1) intercepting the disease at an earlier stage of cytogenetic damage with the goal of eradicating the tumor or substantially prolonging time to progression, and (2) preventing end-organ damage with the goals of improving quality of life and extending survival.
The ongoing development of novel, more refined molecular markers and imaging technology will improve our ability to discriminate between a stable, asymptomatic condition and a progressive disease. This is particularly relevant for patients with high-risk SMM. Preliminary data from ongoing trials in this patient population indicate that this goal might be reachable.38 The PETHEMA group is conducting a multicenter randomized clinical trial designed to assess time-to-progression to symptomatic MM, and efficacy and toxicity of a lenalidomide-dexamethasone schedule versus no treatment in patients with high-risk SMM.57 The last update of this study shows that, in high-risk SMM patients, early treatment with lenalidomide-dexamethasone as induction, followed by lenalidomide as maintenance, prolonged time to progression to symptomatic disease significantly, resulting in a benefit in overall survival (P = .04; M. V. Mateos, personal communication).
At present, the 2010 IMWG guidelines offer an efficient and effective risk-adapted strategy that allows us to “let alone,” as advocated by Jan Waldenström, a substantial proportion (40%) of patients with low-risk MGUS and to establish a watchful follow-up for those at high risk so that we may intervene in a timely fashion. However, these guidelines need to be implemented with widely available, sensitive biomarkers of organ (cardiac) damage to detect in a timely manner the progressive injury produced by toxic light chains, which are usually synthesized by small, indolent PC clones.
Acknowledgments
The authors thank Dr Joan Bladé (Department of Hematology, Hospital Clinic, Barcelona, Spain) for critical reading of the manuscript.
This work was supported by the Ministry of Research and University (2007AESFX2-003), and Associazione Italiana per la Ricerca sul Cancro Special Program Molecular Clinical Oncology (grant 9965).
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: lenalidomide in SMM.
Correspondence
Giampaolo Merlini, MD, Amyloidosis Research and Treatment Center, Fondazione IRCCS Policlinico San Matteo, Viale Golgi, 19-27100 Pavia, Italy; Phone: +39-0382-502994; Fax: +39-0382-502990; e-mail: gmerlini@unipv.it.
