
Abstract
The management of chronic lymphocytic leukemia (CLL) is undergoing profound changes. Several new drugs have been approved for CLL treatment (fludarabine, bendamustine, and the monoclonal antibodies alemtuzumab, rituximab, and ofatumumab) and many more drugs are in advanced clinical development to be approved for this disease. In addition, the extreme heterogeneity of the clinical course and our improved ability to foresee the prognosis of this leukemia by the use of clinical, biological, and genetic parameters now allow us to characterize patients with a very mild onset and course, an intermediate prognosis, or a very aggressive course with high-risk leukemia. Therefore, it becomes increasingly challenging to select the right treatment strategy for each condition. This article summarizes the currently available diagnostic and therapeutic tools and gives an integrated recommendation of how to manage CLL in 2013. Moreover, I propose a strategy how we might integrate the novel agents for CLL therapy into sequential treatment approaches in the near future.
Overview
With an age-adjusted incidence of 4.3/100 000 inhabitants in the United States, chronic lymphocytic leukemia (CLL) is the most common type of leukemia in Western countries. More than 15 000 newly diagnosed cases and ∼ 4500 deaths are currently estimated.1 The median age at diagnosis lies between 67 and 72 years. More male than female patients are affected by this disease.1
CLL is characterized by the clonal proliferation and accumulation of mature, typically CD5-positive B cells within the blood, BM, lymph nodes, and spleen. The leukemic transformation is initiated by specific genomic alterations, in particular deletions on the long arm of chromosome 13 [del(13q14)] Some of these aberrations cause the deletion of specific micro-RNA genes and increase the resistance of B cells toward apoptosis.2,3 Additional aberrations of the long arm of chromosome 11 [del(11q)], of the short arm of chromosome 17 [del(17p)], and trisomy 12 seem to occur later in the course of the disease and predict a worse outcome. In addition, whole-genome sequencing has uncovered recurrent somatic gene mutations that occur in CLL cells in parallel to the above-mentioned structural genomic aberrations. Of these, mutations affecting the genes NOTCH1, MYD88, TP53, ATM, and SF3B1 seem to be more common and to have prognostic impact.4-7
It has become increasingly clear that survival of CLL cells is not a cell-autonomous, genetically determined process. Instead, survival of CLL cells strictly depends on a permissive microenvironment composed of cellular components such as macrophages, T cells, or stromal follicular dendritic cells,8 which provide essential proteins (chemokines, cytokines, and angiogenic factors) for activation of crucial survival and proliferative signaling pathways of transformed cells. In addition, stimulation of the BCR also drives the activation of different tyrosine kinases, such as Bruton tyrosine kinase (BTK), spleen tyrosine kinase (Syk), ZAP70, Src family kinases (in particular Lyn kinase), and phosphatidyl inositol 3-kinase (PI3K), which stimulate malignant B-cell survival via activation of transcription factors such as NF-κB.9,10 The importance of BCR signaling is underscored by the fact that different features of the BCR have been recognized as a prognostic marker in CLL, such as the degree of immunoglobulin heavy chain variable gene (IGHV) mutations or IGHV stereotype (for review, see Stevenson et al9 ). These pathways have recently gained in importance because they can be inhibited by specific small-molecule inhibitors that show clinical efficacy in lymphoid malignancies.10 Excellent reviews have recently summarized these BCR-mediated pathways and the corresponding therapeutic inhibitors.10,11 In some contrast, this review intends to provide an update of the current state-of-the-art of CLL therapy and to propose strategies on how to integrate these novel inhibitors into future CLL therapies.
Standard of care using the currently approved agents
Cytostatic agents
Monotherapy with alkylating agents has served as initial, frontline therapy for CLL, and chlorambucil (CLB) was the gold standard of CLL therapy for several decades.12 Even today, this drug remains an appropriate option for frail elderly or unfit patients. The advantages of CLB are its moderate toxicity, low cost, and convenience as an oral drug; the major disadvantages are its low to nonexistent complete remission (CR) rate and some side effects that occur after extended use (eg, prolonged cytopenia, myelodysplasia, and secondary acute leukemia). Novel results indicate that CLB monotherapy may be used less frequently because the combination with anti-CD20 antibodies has proven more effective (see below).
Three purine analogs are currently used in CLL: fludarabine, pentostatin, and cladribine (2-CdA). Fludarabine remains by far the best-studied compound of the 3 in CLL. Fludarabine induced more remissions and more CR (7%-40%) than other conventional chemotherapies such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), CAP (cyclophosphamide, doxorubicin, prednisone), or CLB, but did not improve overall survival (OS) when used as single agent.13-16 Similarly, cladribine monotherapy was shown to produce a higher CR rate than CLB plus prednisone (47% vs 12%) without resulting in a longer survival.17
More recently, bendamustine was compared with CLB in a randomized trial. Bendamustine produced improved responses but greater toxicity and no OS benefit.18 The overall response rate (ORR) and median progression-free survival (PFS) were 67% and 22 months, respectively, for bendamustine versus 30% and 8 months for CLB (both P < .0001).
Antibodies
Rituximab.
In CLL, the anti-CD20 antibody rituximab is less active as a single agent than in follicular lymphoma unless very high doses are used.19,20 In contrast, combinations of rituximab with chemotherapy have proven to be very efficacious therapies for CLL (see below). Some newer CD20 antibodies challenge rituximab.21-23
Ofatumumab.
Ofatumumab is a fully humanized antibody targeting a unique epitope on the CD20 molecule expressed on human B cells, resulting in increased binding affinity to CD20, prolonged dissociation rate, and increased cell kill due to greater complement-dependent cytotoxicity and similar antibody-dependent cellular cytotoxicity activity compared with rituximab, especially in cells expressing low levels of CD20. Ofatumumab has shown some efficacy in patients who are fludarabine and alemtuzumab refractory or who have bulky disease (> 5 cm).24 The ORR was 58% in the fludarabine and alemtuzumab (FA)–refractory group and 47% in the bulky disease group. However, there has been no formal comparative trial of rituximab versus ofatumumab. Therefore, the value of ofatumumab for the treatment of B-cell lymphoma and CLL remains unclear.
Alemtuzumab.
Alemtuzumab is a recombinant, fully humanized monoclonal antibody against the CD52 antigen. Monotherapy with alemtuzumab has produced response rates of 33% to 53%, with a median duration of response ranging from 8.7 to 15.4 months, in patients with advanced CLL who were previously treated with alkylating agents and had failed or relapsed after second-line fludarabine therapy.25-27 In addition, alemtuzumab has proven effective in patients with high-risk genetic markers such as deletions of chromosome 11 or 17 [del(11q) and del(17p)] and TP53 mutations.28,29 Therefore, alemtuzumab is a reasonable therapeutic option for patients with these poor prognostic features. In a prospective, randomized study, alemtuzumab was tested against CLB.30 Alemtuzumab led to a greater ORR and CR (P < .0001), superior PFS with a 42% reduction in risk of progression or death (P < .0001), and significantly longer median time to progression (TTP; P = .0001). Therefore, the drug remains a valuable treatment option for high-risk patients. Unfortunately, the drug is no longer licensed, but remains available in some countries on a compassionate use basis.
Combination chemotherapy
A major advance in CLL treatment was achieved by the combined use of different treatment modalities. Because purine analogs and alkylating agents have different mechanisms of action and partially nonoverlapping toxicity profiles, they were combined both in preclinical and clinical studies. The most thoroughly studied combination chemotherapy for CLL is fludarabine plus cyclophosphamide (FC) given as oral drugs or IV infusions.31 In noncomparative trials, the ORRs did not appear to be better than with fludarabine alone, but the addition of cyclophosphamide appeared to improve the CR rate up to 50%.31 Three randomized trials have shown that FC combination chemotherapy improves the CR and ORR and PFS compared with fludarabine monotherapy.32-34 The rate of severe infections was not significantly increased by the FC combination despite a higher frequency of neutropenias. An updated analysis of the CLL4 trial of the German CLL Study Group (GCLLSG) suggested that the frontline treatment of CLL patients with FC combination might improve the OS of the non-high-risk CLL patients [all patients not exhibiting a del(17p) or TP53 mutation].
The Polish Adult Leukemia Group (PALG) compared 2-CdA alone with 2-CdA combined with cyclophosphamide (CC) or cyclophosphamide and mitoxantrone (CMC) in 479 patients with untreated progressive CLL.35 Surprisingly, the CC combination therapy did not produce any benefit in terms of PFS or response rates compared with 2-CdA alone.
Chemoimmunotherapy
Combinations using rituximab.
Phase 2 studies suggested that rituximab combinations with fludarabine or fludarabine-based regimens would produce significant improvements in all major outcome parameters such as CR rates, PFS, and OS (for review, see Hallek and Pflug36 ). The largest of these trials, conducted on 300 patients with previously untreated CLL, showed that rituximab plus fludarabine/cyclophosphamide (FCR) achieved an ORR of 95%, with CR in 72%.37 Six-year OS and failure-free survival were 77% and 51%, respectively, and median TTP was 80 months.
These results led the GCLLSG to conduct a randomized trial, the CLL8 protocol, on 817 patients, median age 61 years, with good physical fitness who received 6 courses of FC (n = 409) or FCR (n = 408).38 FCR treatment was more frequently associated with Common Terminology Criteria (CTC) grade 3 and 4 neutropenia (FCR 34%; FC 21%), without any formal recommendation to use growth factor support. Other grade 3 or 4 side effects, including infections, were not increased. FCR induced a higher ORR than FC (92.8% vs 85.4%) and more CR (44.5% vs 22.9%; P < .001). Most importantly, this trial was the first to show that the choice of a frontline therapy has an impact on OS of CLL patients. In an updated analysis of at a median follow-up of 5.9 years, 38.0% of the patients in the FCR group were free of disease progression compared with 27.4% in the FC group (hazard ratio [HR] = 0.6; 95% confidence interval [95% CI], 0.5-0.7; P < .0001).39 At the same time point, 69.4% of the patients were alive in the FCR group versus 62.3% in the FC group (HR = 0.7; 95% CI, 0.5-0.9), P = .001). Median OS was not reached for patients in the FCR group, whereas the median OS was 86.0 months (95% CI, 78.7-93.2 months) for the FC group (P = .001).39 FCR did not improve the survival of patients with a del(17p). Similar results were obtained in a trial comparing FCR without FC in relapsed CLL without demonstrating a benefit for OS.40
Because CLL often occurs in elderly patients with relevant comorbidity, a dose-modified FCR-Lite regimen was designed to maintain the efficacy but decrease the toxicity of the FCR regimen.41 This regimen reduced the dose of fludarabine and cyclophosphamide, increased the dose of rituximab, and used a maintenance regimen for rituximab given every 3 months until progression. The CR rate was 77% for 50 previously untreated CLL patients with an ORR of 100%. Grade 3/4 neutropenia was documented in only 13% of cycles, which is lower than that observed with the usual FCR regimen.
Following a similar concept, the CLL11 protocol of the GCLLSG was designed to test chemoimmunotherapies with anti-CD20 antibodies combined with a milder chemotherapeutic component, CLB, in previously untreated CLL patients with comorbidities.42 The rationale of this study was based on encouraging results obtained in phase 2 trials using CLB in combination with rituximab (RCLB).43,44 The CLL11 trial compared CLB alone versus a new anti-CD20 antibody, obinutuzumab (GA101; see below), plus CLB (GCLB) versus rituximab (R) plus CLB (RCLB). The study included treatment-naive CLL patients with a Cumulative Illness Rating Scale (CIRS) total score > 6 and/or an estimated creatinine clearance (CrCl) < 70 mL/min. Treatment consisted of CLB alone (0.5 mg/kg orally on days 1 and 15 for 28 days and 6 cycles), GCLB (100 mg IV on day 1, 900 mg on day 2, 1000 mg on days 8 and 15 of cycle 1, 1000 mg on day 1 for cycles 2-6), or RCLB (375 mg/m2 IV on day 1 cycle 1, 500 mg/m2 on day 1 cycles 2-6).42 The median age of patients was 73 years and the median CIRS score was 8. So far, the results obtained allow the conclusion that chemoimmunotherapy with one of the 2 anti-CD20 antibodies increases the ORR, allows patients to achieve CRs (22% vs 8% vs 0%; GCLB vs RCLB vs CLB) and seems to prolong PFS from 11 months (CLB) to 16 or 23 months (RCLB or GCLB), respectively. The major pronounced side effects were grade 3-4 infusion-related reactions with GCLB at the first infusion. Taken together, chemoimmunotherapy with anti-CD20 antibodies is a potent treatment concept in CLL patients with comorbidities as well.
Other variations have been tested to further improve the efficacy of the FCR regimen: Alemtuzumab (A) was added to FCR (CFAR) in a phase 2 trial on 60 high-risk untreated patients < 70 years of age with serum beta2-microglobulin ≥4 mg/L.45 CR was achieved in 70%, PR in 18%, nodular PR in 3%, for an ORR of 92%. Of 14 patients with 17p deletion, 8 (57%) achieved a CR. Grade 3/4 neutropenia and thrombocytopenia occurred with 33% and 13% courses, respectively. The median PFS was 38 months and median OS was not reached. Therefore, CFAR might be helpful in cases of high-risk CLL in which an effective cytoreductive therapy is desired before an allogeneic stem cell transplantation. In another study on 72 untreated CLL patients ≤70 years of age, mitoxantrone was combined at 6 mg/m2 on day 1 of each cycle with FCR.46 The ORR, minimal residual disease (MRD)–negative CR, MRD-positive CR, and PR rates were 93%, 46%, 36%, and 11%, respectively. Severe neutropenia developed in 13% of patients. These results do not justify the broad use of this regimen outside of clinical trials.
An alternative idea was to replace fludarabine in the FCR regimen with pentostatin (PCR) to reduce myelotoxicity. In a phase 3 randomized trial comparing FCR with PCR in previously untreated or minimally treated CLL patients, there were no statistical differences between treatments in OS or response rates.47 Moreover, this trial did not demonstrate a lower infection rate with PCR.
Bendamustine has been also combined with rituximab (BR) in 81 patients with relapsed CLL.48 Patients received 70 mg/m2 of bendamustine on days 1 and 2 and 375 mg/m2 of rituximab on day 0 of the first cycle and 500 mg/m2 on day 1 of subsequent cycles administered every 28 days for up to 6 cycles. On the basis of intent-to-treat analysis, the ORR was 59.0% (95% CI, 47.3%-70.0%). CR, PR, and nodular PR were achieved in 9.0%, 47.4%, and 2.6% of patients, respectively. ORR was 45.5% in fludarabine-refractory patients and 60.5% in fludarabine-sensitive patients. Among genetic subgroups, 92.3% of patients with del(11q), 100% with trisomy 12, 7.1% with del(17p), and 58.7% with unmutated IGHV status responded to treatment. After a median follow-up time of 24 months, the median event-free survival was 14.7 months. Severe infections occurred in 12.8% of patients. Grade 3 or 4 neutropenia, thrombocytopenia, and anemia were documented in 23.1%, 28.2%, and 16.6% of patients, respectively.
The BR regimen was also investigated as frontline therapy in 117 CLL patients (age 34-78 years).49 Bendamustine was administered at a dose of 90 mg/m2 on days 1 and 2 combined with 375 mg/m2 rituximab on day 0 of the first course and 500 mg/m2 on day 1 during subsequent courses for up to 6 courses. ORR was 88.0% (95% CI, 80.7%-100.0%) with a CR rate of 23.1% and a PR rate of 64.9%. Ninety percent of patients with del(11q), 94.7% with trisomy 12, 37.5% with del(17p), and 89.4% with unmutated IGHV status responded to treatment. After a median observation time of 27.0 months, median event-free survival was 33.9 months, and 90.5% of patients were alive. Grade 3 or 4 severe infections occurred in 7.7% of patients. Grade 3 or 4 adverse events for neutropenia, thrombocytopenia, and anemia were documented in 19.7%, 22.2%, and 19.7% of patients, respectively. Overall, these results suggest that, compared with FCR, BR is somewhat less active, yielding lower CR rates, but is also less myelotoxic. The GCLLSG is currently comparing BR with FCR in a randomized phase 3 trial, the CLL10 protocol.
Several other combinations have been investigated, such as cladribine with rituximab, methylprednisolone plus rituximab followed by alemtuzumab, or rituximab plus alemtuzumab. Their detailed description is beyond the scope of this article, because none of them has been proven to result in higher efficacy compared with FCR.
Combinations using alemtuzumab.
The synergistic activity of fludarabine and alemtuzumab was initially suggested by the induction of responses, including 1 CR, in 5 of 6 patients who were refractory to each agent alone.50 The combination of FA was investigated in a phase 2 trial enrolling patients with relapsed CLL using a 4-weekly dosing protocol.51 This combination has proven feasible, safe, and effective. Among the 36 patients, the ORR was 83% (30/36 patients), which included 11 CRs (30%) and 19 PRs (53%); there was one stable disease. Sixteen of 31 evaluated patients (53%) achieved MRD negativity in the peripheral blood by 3 months of follow-up. Resolution of disease was observed in all disease sites, particularly in the blood, BM, and spleen. The FA therapy was well tolerated. Infusion reactions (fever, chills, and skin reactions) occurred primarily during the first infusions of alemtuzumab and were mild in the majority of patients. Although 80% of patients were CMV IgG-positive before treatment, there were only 2 subclinical CMV reactivations. The primary grade 3/4 hematological events were transient, including leukocytopenia (44%) and thrombocytopenia (30%). Stable CD4+ T-cell counts (> 200/μl) were seen after 1 year.
Two phase 3 trials tested alemtuzumab in combination with FC (FCA) or fludarabine (FA). One trial comparing FCA with FCR in frontline therapy was closed prematurely due to the higher toxicity and treatment-related mortality observed in the FCA arm.52 The therapeutic efficacy of FCR was clearly superior to FCA. In this trial, alemtuzumab was given subcutaneously. A second randomized trial compared FA with fludarabine monotherapy in previously treated patients with relapsed or refractory CLL.53 In this trial, alemtuzumab was given IV. FA (n = 168) resulted in better PFS than fludarabine monotherapy (n = 167; median 23.7 months vs 16.5 months; HR = 0.61; P = .0003) and OS (median not reached vs 52.9 months; HR = 0.65; P = .021) compared with fludarabine alone. Adverse events occurred in 161 (98%) of 164 patients in the FA group and 149 (90%) of 165 in the fludarabine alone group. Patients in the FA group had more CMV events (14% vs < 1%) and grade 1 or 2 infusion-related adverse reactions (62% vs 13%). Major grade 3 or 4 toxicities in the FA and monotherapy groups were leukopenia (74% vs 34%), lymphopenia (94% vs 33%), neutropenia (59% vs 68%), thrombocytopenia (11% vs 17%), and anemia (9% vs 17%). The incidence of serious adverse events was higher in the FA group (33% vs 25%); deaths due to adverse events were similar between the 2 groups (6% vs 12%).
Two phase 2 trials investigated alemtuzumab in combination with high-dose corticosteroids, which seem to be a good therapeutic option, in particular for patients with del(17p). The combination of alemtuzumab and methylprednisolone was tested in the UK CLL206 trial on 17 untreated and 22 previously treated patients with del(17p). The ORR and CR rate were 85% and 36% in the whole cohort and 88% and 65% in treatment naive patient group, respectively.54 In the CLL2O trial of the GCLLSG, the combination of alemtuzumab and high-dose dexamethasone resulted in an ORR above 90% in treatment-naive high-risk patients.55
Selecting the right treatment: how to treat CLL with the currently approved agents?
Given the impressive number of novel drugs, the right choice of treatment for a given CLL patient has become a task that requires experience, a good clinical assessment of the patient, and an appropriate use of diagnostic tools. The following parameters should be considered before recommending a treatment for CLL56 : (1) the clinical stage of disease, (2) the fitness of the patient, (3) the genetic risk of the leukemia, and (4) the treatment situation (frontline vs second-line, response vs nonresponse of the last treatment). With the use of these 4 parameters, the following recommendations can be given (Figure 1A,B):

Treatment algorithm for CLL patients in frontline (A) and second-line (B) indications. Al indicates alemtuzumab; R, rituximab; O, ofatumumab; F, fludarabine; C, cyclophosphamide; Mab, monoclonal antibody; Dex, dexamethasone; and Allo-SCT, allogeneic stem cell transplantation. Please note that performing an allogeneic transplantation usually requires the induction of a PR or CR before the procedure.
Treatment algorithm for CLL patients in frontline (A) and second-line (B) indications. Al indicates alemtuzumab; R, rituximab; O, ofatumumab; F, fludarabine; C, cyclophosphamide; Mab, monoclonal antibody; Dex, dexamethasone; and Allo-SCT, allogeneic stem cell transplantation. Please note that performing an allogeneic transplantation usually requires the induction of a PR or CR before the procedure.
Frontline treatment (Figure 1A).
In a patient with advanced (Binet C, Rai III-IV) or active, symptomatic disease, treatment should be initiated. In this situation, patients need to be evaluated for their physical condition (or comorbidity). For patients in good physical condition (“go go”), as defined by a normal creatinine clearance and a low score at the “cumulative illness rating scale” (CIRS),57 patients should be offered chemoimmunotherapies such as FCR or FR to achieve sustained remissions.
Patients with a somewhat impaired physical condition (“slow go”) may be offered either CLB in combination with an anti-CD20 antibody42 or a dose-reduced fludarabine-containing regimen with a CD20 antibody. The aim of therapy in this situation is symptom control.
Patients with symptomatic disease and with del(17p) or TP53 mutations may receive FCR or BR or an alemtuzumab-containing regimen as frontline treatment. In general, these regimens all yield response rates above 50%, but the TTP tends to be shorter than 2 years. The recent results obtained with single-agent ibrutinib in refractory or relapsed patients with a del(17p) showed an ORR of 68%, with a PFS at 26 months of 57% and an OS of 70%.58 Although these results appear very encouraging, none of these therapies promises long-lasting remissions. Therefore, allogeneic stem cell transplantation should still be offered and discussed in patients with sufficient physical fitness.
Second-line treatment (Figure 1B).
As a general rule, the frontline treatment may be repeated if the duration of the first remission exceeds 24 months provided that the frontline therapy was well tolerated. The choice becomes more difficult and limited in treatment-refractory CLL, in patients relapsing within 24 months of treatment, or in patients with the chromosomal aberration del(17p). In principle, the initial regimen should be changed. The following options exist as a relapse therapy for patients no longer responding to chemoimmunotherapy with rituximab: (1) alemtuzumab alone or in combinations, in particular with high-dose steroids26,51,54 ; (2) combinations of antibodies with lenalidomide (see below); (3) BTK inhibitors such as ibrutinib,58 where available, or experimental protocols using novel agents (Figure 2, Table 1); and (4) allogeneic stem cell transplantation with curative intent.59

Targeting of BCR signaling as a therapeutic strategy in CLL. Red symbols and letters indicate new therapeutics as discussed in the text.
Targeting of BCR signaling as a therapeutic strategy in CLL. Red symbols and letters indicate new therapeutics as discussed in the text.
Status of current clinical development of novel, targeted drugs in CLL
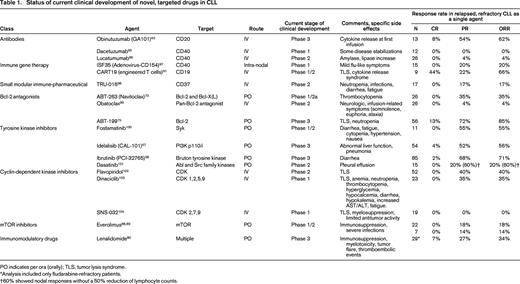
PO indicates per ora (orally); TLS, tumor lysis syndrome.
*Analysis included only fludarabine-refractory patients.
†60% showed nodal responses without a 50% reduction of lymphocyte counts.
The choice of one of these options depends on the fitness of the patient, the availability of some drugs, and the prognostic risk of the leukemia as defined by molecular cytogenetics. According to recommendations of a European Group for Blood and Marrow Transplantation (EBMT) consensus group, physically fit patients with refractory CLL or with a del(17p) should be offered an allogeneic transplantation because their prognosis so far has remained extremely poor with conventional therapies.59 Finally, it is important to emphasize that patients with refractory disease should be treated within clinical trials whenever possible. Some of the new drugs, in particular ibrutinib or ABT-199, show good responses in these patients and could become an additional option in the very near future.
New drugs targeting pathogenic pathways of CLL cells
There are an increasing number of interesting new compounds in clinical development (for reviews, see Wiestner10 and Isfort et al60 ). The common denominator of these compounds is that their mechanism of action targets a relatively specific signaling abnormality or redirects the immune system against CLL cells. A detailed description of these fascinating agents is beyond the scope of this article. Table 1 lists the some of the most promising agents and their stage of clinical development and shows that these agents may yield high response rates above 50% even in relapsed and refractory CLL patients. Some of these agents (eg, obinutuzumab, CART19, ABT-199, idelalisib, and ibrutinib) currently elicit the greatest enthusiasm and hope both among CLL patients and their treating physicians.
New anti-CD20 antibodies
Obinutuzumab (GA101).
The humanized and glycoengineered monoclonal antibody obinutuzumab showed impressive results in vitro with higher rates of apoptosis in malignant B cells compared with rituximab.61 The humanization of the parental B-Ly1 mouse antibody and subsequent glycoengineering lead to higher-affinity binding to the CD20 type II epitope, increased antibody-dependent cellular cytotoxicity, low complement-dependent cytotoxicity activity, and increased direct cell death induction.62 A phase 1 study with obinutuzumab showed promising results in 13 CLL patients.63 Major side effects included infections, neutropenia, thrombocytopenia, and tumor lysis syndrome, all of which resolved. There were no dose-limiting toxicities. Encouraging results were reported in the run-in phase of the CLL11 trial on CLL patients with increased comorbidity.64 Six subjects older than 70 years (median age 76 years; median cumulative illness rating score of 8) received 6 28-day cycles of obinutuzumab (1000 mg on days 1, 8, and 15 of cycle 1 and day 1 of cycles 2-6) plus CLB (0.5 mg/kg on days 1 and 15 of each cycle). Infusion-related reactions occurred in 5 patients, but were mild. Grade 3/4 neutropenia was seen in 5 patients. No febrile neutropenias or grade 3/4 infections were observed. All subjects completed therapy. CRs were documented in 2 patients and PRs in 4. After a median follow-up of 23 months from start of treatment, none of the patients had progressed with CLL or died. Results of a planned interim analysis of the CLL11 trial have been recently reported and confirmed the very promising activity of obinutuzumab in CLL.42
Agents targeting BCR signaling
The following section describes selected kinase inhibitors that have entered testing in phase 3 trials for CLL patients (Table 1).
Idelalisib (CAL-101).
Class I PI3Ks regulate cellular functions relevant to oncogenesis.65 Expression of the PI3K p110 δ isoform (PI3Kδ) is restricted to cells of hematopoietic origin, where it plays a key role in B-cell proliferation and survival. In CLL, the PI3K pathway is constitutively activated and dependent on PI3Kδ.66 CAL-101 is an oral PI3Kδ isoform–selective inhibitor that promotes apoptosis in primary CLL cells in a time- and dose-dependent manner without inducing apoptosis in normal T cells or natural killer cells and without diminishing antibody-dependent cellular cytotoxicity. CAL-101 inhibits CLL cell chemotaxis toward CXCL12 and CXCL13 and migration beneath stromal cells (pseudoemperipolesis). CAL-101 also down-regulates the secretion of chemokines in stromal cocultures and after BCR triggering.66 CAL-101 reduces survival signals derived from the BCR or from nurse-like cells and inhibits BCR- and chemokine-receptor-induced AKT and MAP kinase (ERK) activation.66
In a phase 1 clinical trial in 54 heavily pretreated and high-risk CLL patients, idelalisib showed acceptable toxicity, positive pharmacodynamic effects, and favorable clinical activity (ie, a high level of lymph node regression and prolonged duration of symptomatic tumor control).67 An ORR of 56% was achieved, with 2 CRs and 28 PRs. Of the 28 patients with PRs, 6 showed persistent lymphocytosis. The majority of patients (81%) showed a lymph node response (≥ 50% reduction in the nodal sum of the product of greatest diameter). Median PFS was 17 months. Side effects were mild, with fatigue, diarrhea, pyrexia, rash, and upper respiratory tract infections being the most frequent. More importantly, there were no dose-limiting toxicities. Results on idelalisib in combination with rituximab, ofatumumab, or bendamustine/rituximab were presented in preliminary form and showed encouraging results. For example, a trial using a combination of idelalisib and rituximab as a frontline therapy in 64 patients yielded an ORR of 97% with 19% CRs.68 Side effects were mild, with 23% diarrheas as the only relevant CTC grade 3 toxicity.
Ibrutinib (formerly called PCI-32765).
BTK leads to downstream activation of cell survival pathways such as NF-κB and MAPKs via Src family kinases.69 Ibrutinib is an orally active small molecule inhibiting BTK that plays a role in the signal transduction of the BCR. Inhibition of BTK might induce apoptosis in B-cell lymphomas and CLL cells.69 Ibrutinib showed significant activity in patients with relapsed or refractory B-cell malignancies, including CLL.70 Data from a phase 1b-2 multicenter study with single-agent ibrutinib on 85 patients with relapsed or refractory CLL or small lymphocytic lymphoma, the majority of whom had high-risk disease, were published recently.58 Patients received ibrutinib once daily (51 at a dose of 420 mg, 34 at 840 mg). Side effects were mild (predominantly grade 1 or 2) and included transient diarrhea, fatigue, and upper respiratory tract infection. The ORR was 71%, the majority being PRs (68%). Most interestingly, the response was independent of clinical and genomic risk factors, including advanced-stage disease, the number of previous therapies, and del(17p). At 26 months, the estimated PFS rate was 75% and the OS rate was 83%. This study illustrates that ibrutinib may soon become an additional treatment option for CLL patients with high-risk genetic lesions.
Bcl-2 inhibitors
Proteins in the B-cell CLL/lymphoma 2 (Bcl-2) family are key regulators of the apoptotic process.71 The Bcl-2 family comprises proapoptotic and prosurvival proteins. Shifting the balance toward the latter is an established mechanism whereby cancer cells evade apoptosis. Bcl-2, the founding member of this protein family, is encoded by the BCL2 gene, which was initially described in follicular lymphoma as a protein in translocations involving chromosomes 14 and 18.72
Bcl-2 inhibitors ABT-263 (Navitoclax) and ABT-199.
ABT-263 is a small-molecule Bcl-2 family protein inhibitor that binds with high affinity (Ki ≤ 1 nM) to multiple antiapoptotic Bcl-2 family proteins, including Bcl-XL, Bcl-2, Bcl-w, and Bcl-B, and has a high oral bioavailability. Initial studies showed very promising results for this drug as a single agent.73 However, its therapeutic use seemed somewhat limited by severe thrombocytopenias being a prominent side effect. Therefore, the compound was reengineered to create a highly potent, orally bioavailable, and Bcl-2-selective inhibitor, ABT-199.74 This compound inhibits the growth of Bcl-2–dependent tumors in vivo and spares human platelets. A single dose of ABT-199 in 3 patients with refractory CLL resulted in tumor lysis within 24 hours.74 In a recent update on phase 1 study data of 56 patients have been reported, of whom 16 (29%) had a del(17p) and 18 (32%) had F-refractory CLL.75 Major side effects were tumor lysis syndrome and neutropenia. ABT-199 yielded an ORR of 85%, with 13% CRs and 72% PRs. Interestingly, 88% and 75% of patients with a del(17p) and F-refractory CLL, respectively, achieved at least a PR. These data indicate that selective pharmacological inhibition of Bcl-2 holds great promise for the treatment of CLL.
Immunomodulatory drugs
Lenalidomide.
Lenalidomide has shown encouraging results in the treatment of high-risk patients, including carriers of a del(17p).76 In 58% of the patients, lenalidomide caused a tumor flare reaction characterized by a marked and painful increase in lymph nodes size, malaise, and fever.77,78 This tumor flare reaction may be life-threatening and is more common in CLL than in other lymphoid malignancies. Lenalidomide may also cause relevant myelosuppression.79 The ORR of lenalidomide monotherapy varied between 32% and 54% in different clinical trials.78-80 Most importantly, it seems to have activity as a single agent in fludarabine-refractory CLL.78,80
The combination of lenalidomide and rituximab seems to increase the response rate without a higher risk of toxicity, even in patients with del(17p) and/or unmutated IGHV status. In a phase 2 trial, 59 patients with relapsed or refractory CLL received a combination of lenalidomide and rituximab.81 Lenalidomide was started on day 9 of cycle 1 at 10 mg orally and was administered daily continuously. Each cycle was 28 days. Rituximab was administered for 12 cycles; lenalidomide could be continued if patients benefited clinically. The ORR was 66%, including 12% CRs and 12% nodular PRs. The median time to treatment failure was 17.4 months. The most common grade 3 or 4 toxicity was neutropenia (73% of patients). Fourteen patients (24%) experienced a grade 3 or 4 infection or febrile episode. In essence, this combination seems a helpful alternative for patients with refractory CLL and warrants further investigation.
In some contrast, the combination of lenalidomide, rituximab, and fludarabine may induce severe side effects (myelosuppression) if all drugs are started simultaneously on day 1.82,83 Using combinations starting with lenalidomide on day 8 of the first cycle, the treatment is usually better tolerated.83,84 The GCLLSG is currently investigating the combination of bendamustine, rituximab, and lenalidomide (BR2) in physically fit patients (CLL2P protocol). Additional combinations currently being studied are flavopiridol plus lenalidomide, which led to a response in 7 of 15 patients [among them 4 with a del(17p) and 3 with a del(11q)]85 or lenalidomide plus ofatumumab.86
Everolimus (RAD001).
Chimeric antigen receptors
Chimeric antigen receptors (CARs) combine the antigen recognition domain of an antibody with intracellular signaling domains into a single chimeric protein. CD19 is an ideal target for CARs because expression is restricted to normal and malignant B cells. Inclusion of the CD137 (4-1BB) signaling domain resulted in potent antitumor activity and in vivo persistence of anti-CD19 CARs in mice. June et al have recently shown that antitumor activity of CAR-modified autologous T cells targeted to CD19 (CART19 cells) yielded sustained responses even in high-risk CLL patients.90 Recently, the outcome and longer follow-up from 10 patients treated with CART19 cells was reported.91 Autologous T cells collected by leukapheresis were transduced with a lentivirus encoding anti-CD19 scFv linked to 4-1BB and CD3-z signaling domains. Gene-modified T cells were expanded and activated ex vivo by exposure to anti-CD3/CD28 beads. Ten patients have received CART19 cells; 9 adults of median age 65 years (range, 51-78) were treated for relapsed, refractory CLL and 1 7-year-old was treated for relapsed refractory acute lymphoblastic leukemia (ALL). Three of 9 CLL patients had a deletion of the p53 gene. All CLL patients received lymphodepleting chemotherapy 4 to 6 days before infusions (FC, PC, or bendamustine, whereas the ALL patient had an absolute lymphocyte count < 10 after prior chemotherapy and did not require further lymphodepletion). CART19 homed to the BM in the CLL patients and in the BM and CSF in the ALL patient, with detectable CART19 cells in the CSF (21 lymphocytes/μL, 78% CAR+) day 23 after infusion. Four of 9 evaluable patients achieved a CR (3 CLL, 1 ALL). Two CLL patients had a PR lasting 3 and 5 months and 3 patients did not respond. In the 4 patients who achieved CR, maximal expanded cells in the blood were detected at an average of 27-fold higher than the infused dose (range, 21- to 40-fold) with maximal in vivo expansion between day 10 and 31 after infusion. No patient with CR has relapsed. All patients who responded developed a cytokine release syndrome manifested by fever and variable degrees of nausea, anorexia, and transient hypotension and hypoxia. In responding CLL patients, cytokine levels were increased. Five patients with cytokine release required treatment. In summary, CART19 cells can induce potent and sustained responses for patients with advanced, refractory, and high-risk CLL. However, the long-term toxicity and efficacy of this approach needs to be studied further.
Outlook: signaling the end of CLL?
The above described, novel therapies all target relatively specific signaling proteins of CLL cells and their microenvironment. Therefore, their overall toxicity often is moderate and does not involve myelosuppression. Moreover, CRs have not occurred frequently so far with any of these newer agents, except CART19.91 In addition, unlike chronic myeloid leukemia, which is initiated by a single oncogene (BCR/ABL), CLL appears to be a genetically heterogeneous leukemia. It seems that CLL may be caused by a complex array of genetic events and as a consequence is a biologically complex disease. Finally, somatic mutations in the kinase genome are very rare events in CLL,92 unlike in other malignancies. For these reasons, it is very likely that the currently available therapeutic agents will be combined to yield optimal results. The current challenge is to identify the best combination and sequence of treatments to achieve the long-term control of CLL with optimal quality of life. Finally, it is important to keep in mind that, as in other hematological malignancies, the currently available data strongly suggest that achieving a very good disease control (using MRD negativity as a surrogate parameter) prolongs survival of CLL patients.38,93
How can these goals be achieved with the current treatment modalities?
In principle there are at least 3 options. The first option is combining the best agents (3, 4, or more) in a simultaneous short-term treatment (up to 6 months) aimed at MRD-negative CRs93 that would last longer than the remissions achieved so far. For example, such an approach would be combining the best currently available chemotherapies (fludarabine or cyclophosphamide alone or in combination; bendamustine) with antibodies and with kinase inhibitors or Bcl-2 antagonists. However, these combinations will have to be compared against the current standard (FCR) and results will not be available for a few years. Moreover, such a treatment strategy will not be without toxicity and therefore will be tolerated only by patients with good physical fitness.
A second possibility would be to use sequential monotherapies of new or old agents. Each agent would be given until maximal response was achieved. After a long-lasting response, treatment could be repeated with the same agent, whereas alternative agents would be used in case of short remissions. This strategy might be applied in elderly or nonfit patients (“slow go”), in whom the goal of treatment is symptom control rather than disease control.
A third strategy would combine the best agents in a sequence that tailors the treatment according to the initial tumor load and the response to therapy (CR vs PR; MRD-positive vs MRD-negative).93 This strategy would have the goal of preventing the outgrowth of adverse leukemic subclones94 and minimizing the use of chemotherapy, thereby reducing the risk for secondary mutations of the CLL clone(s) and for secondary malignancies that are frequent and prognostically unfavorable events in CLL. For this treatment approach, I propose the term “sequential TTT (triple-T)” (tailored, targeted, total eradication of MRD; Figure 3). This sequential triple-T approach will make use of all of the currently available options in a nonaggressive, nontoxic way and will aim at the complete elimination or control of the malignant clone. The advantage of such an approach would be as follows: (1) it would be available for most (fit as well as nonfit) CLL patients due to its limited toxicity, (2) it could be given in an outpatient setting, and (3) it would use the current treatment options in a tailored and response-adjusted manner and therefore use the drugs in a cost-effective, resource-saving way (Figure 3). It might also be used to monitor the evolution of new genetic subclones in CLL that may have clinical and prognostic impact.94

Future treatment concept with novel agents. For this treatment approach, I propose the term “sequential triple-T” (tailored, targeted, total eradication of MRD) to illustrate that this future approach should be a sequence of tailored measures (according to the risk of the leukemia, the tumor burden, and the fitness of the patients), should use targeted agents (ie, using the novel nonchemotherapeutic agents with a mechanism of action targeting pathogenic signaling events of CLL cells and their microenvironment), and aim at the total eradication of the leukemic clone (as assessed by MRD negativity as a clinical end point). Please note that the drugs or classes of drugs in this figure are shown as examples. Similar agents of the same class or additional classes of drugs (see Table 1) may be used as well.
Future treatment concept with novel agents. For this treatment approach, I propose the term “sequential triple-T” (tailored, targeted, total eradication of MRD) to illustrate that this future approach should be a sequence of tailored measures (according to the risk of the leukemia, the tumor burden, and the fitness of the patients), should use targeted agents (ie, using the novel nonchemotherapeutic agents with a mechanism of action targeting pathogenic signaling events of CLL cells and their microenvironment), and aim at the total eradication of the leukemic clone (as assessed by MRD negativity as a clinical end point). Please note that the drugs or classes of drugs in this figure are shown as examples. Similar agents of the same class or additional classes of drugs (see Table 1) may be used as well.
The proposed sequential triple-T strategy might consist of the following 3 steps (Figure 3):
1. Debulking.
Because most of the new agents either induce a compartment shift of malignant lymphocytes (eg, ibrutinib, idelalisib), they transiently increase peripheral blood lymphocyte counts. This often causes concern in patients and physicians and prevents combination with some other drugs. Other agents (ABT-199, obinutuzumab, lenalidomide) often cause severe reactions during the first treatment phase (cytokine release syndrome, tumor lysis syndrome). Therefore, in cases with elevated lymphocyte counts (above 30 000/μl) or large lymph nodes, a short debulking therapy (eg, with 1 or 2 courses of bendamustine or fludarabine) might be an optional first treatment element. Alternatively, nonchemotherapeutic drugs could be used. This step would be performed to rapidly clear the peripheral blood of CLL cells in the majority of patients. This treatment period would last 1 to 2 months.
2. Induction.
After reducing peripheral blood lymphocytes to levels below a certain threshold (eg, below 30 000 peripheral blood lymphocytes/μl), induction therapies could be initiated. These therapies would contain nonchemotherapy combinations of drugs. To avoid infusion-related reactions, antibodies such as obinutuzumab would be given first, followed a few days later by a third class of drugs, such as a tyrosine kinase inhibitor or Bcl-2 inhibitor (or both). This treatment period would typically last 4 to 6 months and the patient would be monitored at the end by MRD assessment 3 months after a CR is achieved. In general, this combination treatment during induction would last as long as the remission continues to improve or until a CR is achieved.
3. Maintenance.
At the end of this induction period, the third sequential element of the triple-T strategy will ensure that a very good remission is maintained. This could be achieved by giving single agents, either oral drugs (eg, kinase inhibitors, Bcl-2 inhibitors, or lenalidomide) or repetitive infusions of antibodies (eg, obinutuzumab) in intervals of 2 to 6 months for a prolonged period. This therapeutic approach would be monitored by MRD assessment and treatment could be stopped 3 months after an MRD-negative remission has been achieved and restarted in case of MRD positivity. This treatment phase would last at least 1 year, but typically 2 years.
Although I am fully aware that the proposed sequential triple-T strategy needs to be validated by clinical research, it may help to design the appropriate clinical trials for the optimal use of currently available drugs. Finally, at a time when new and exciting therapeutic options are likely to change the management and outcome of CLL, hematologists and oncologists have the obligation to include their patients in clinical trials to ensure that maximal progress is achieved in the shortest possible time. By doing so, we will actively contribute to the historically unique chance to gain control over a so-far incurable disease such as CLL.
This article was selected by the Blood and Hematology 2013 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2013. This article is reprinted with permission from Blood. 2013; Volume 122.
Acknowledgments
This work is supported by the Deutsche Krebshilfe (German Cancer Aid), the Kompetenznetz Maligne Lymphome (Competence Network Malignant Lymphoma), and the Deutsche Forschungsgemeinschaft (grants KFO 286 and SFB 832 and Center of Excellence grant “Cellular Stress Responses in Aging-Associated Diseases”). The author thanks Drs Anja Engelke, Gabor Kovacs, and Paula Cramer for carefully reading the manuscript; the team of the GCLLSG study office for giving important suggestions; Dr Günter Krause for generating Figure 2; and all of the patients and physicians participating in the studies of the GCLLSG for their continuing support and excellent cooperation.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Celgene, Janssen, Gilead, Mundipharma, and Roche and has consulted for Mundipharma, Roche, Celgene, Gilead, Janssen, and GSK. Off-label drug use: ibrutinib, cal-101 (idelalisib), obinutuzumab.
Correspondence
Michael Hallek, MD, Kerpener Strasse 62, 50937 Köln, Germany; Phone: 49-221/478-4400; Fax: 49-221/478-5455; e-mail: michael.hallek@uni-koeln.de.
