
Abstract
With the approval in many countries of nilotinib and dasatinib for frontline therapy in chronic myeloid leukemia, clinicians now have to make a difficult choice. Because none of the 3 available tyrosine kinase inhibitors (TKIs) have shown a clear survival advantage, they all represent reasonable choices. However, in individual patients, the case may be stronger for a particular TKI. In the younger patient, in whom the prospect of eventually achieving treatment-free remission is likely to be of great importance, dasatinib or nilotinib may be preferred, although their advantage over imatinib in this setting remains to be proven. In patients with a higher risk of transformation (which is currently based on prognostic scoring), the more potent TKIs may be preferred because they appear to be more effective at reducing the risk of transformation to BC. However, imatinib still represents an excellent choice for many chronic myeloid leukemia patients. All of these considerations need to be made in the context of the patient's comorbidities, which may lead to one or more TKIs being ruled out of contention. Whatever first choice of TKI is made, treatment failure or intolerance must be recognized early because a prompt switch to another TKI likely provides the best chance of achieving optimal response.
The current scene in chronic myeloid leukemia
When a newly diagnosed chronic-phase chronic myeloid leukemia (CP-CML) patients first presents, the clinician is faced with difficult, but welcome, choices. What is the best therapeutic approach for this particular patient given the excellent therapies available? Assuming that the tyrosine kinase inhibitors (TKIs) imatinib, nilotinib, and dasatinib are locally approved, which they are in many countries today, it is unlikely and probably undesirable that the clinician would select the same drug in every case. Although in many cases it makes good sense to use the TKI that the clinician is most familiar with, there is emerging evidence that in certain circumstances, one TKI may be a better choice for a particular patient. Detailed knowledge of the safety profile and efficacy of each TKI, the patient's comorbidities, and clarity about the therapeutic goals will assist drug selection. Before examining the decision process, it is worthwhile reviewing the strengths and weaknesses of the 3 most commonly used frontline TKIs.
Imatinib
There are several hundred thousand patients on imatinib therapy globally, and many patients who have had more than a decade of exposure to this remarkable drug. What is clear is that it is a highly effective TKI for ∼60% of CML patients, poorly tolerated in ∼20%, and inadequate to achieve or maintain an optimal response in a further 20%.1 One of the major drawbacks for imatinib is that it is significantly less potent than nilotinib and dasatinib. Evidence for this comes from in vitro studies and the clinical observation that inadequate kinase inhibition in patients receiving imatinib is not uncommon and is associated with inferior response.2,3 Failure to achieve adequate blood levels is one example. Plasma trough drug levels <1000 ng/mL are associated with inferior response.4,5 However, this is not the only means by which imatinib-induced kinase inhibition in leukemic cells can be inadequate. Susceptibility to inadequate cellular uptake even in the presence of adequate drug levels is due mainly to reduced activity of the OCT-1 influx pump.6,7 The activity of this pump is highly variable and the majority of patients who fail to achieve optimal response to imatinib have evidence of low activity, as measured by the OCT-1 activity assay.8,9 Another major drawback of imatinib as a frontline drug in CP-CML is the frequency of kinase domain mutations that emerge on imatinib therapy.10 This became evident in the ENESTnd study, in which the number of mutations detected on the imatinib arm was twice as high as those on the nilotinib arm (7% vs 3.5%).11 The third significant problem with imatinib is that nearly all patients experience some impairment in quality of life, such as excessive fluid retention, muscle pains and cramps, or gastrointestinal disturbances.12
On the positive side, there are virtually no worrying organ toxicity signals emerging after prolonged exposure to imatinib. The early concerns about an increase in cancer susceptibility and cardiac dysfunction have not been borne out. The other potential advantage is the imminent availability of generic imatinib in many countries, which could potentially greatly ease the cost burden of long-term therapy for CML.
The IRIS trial showed an 8-year survival for imatinib recipients of 85%, 93% if only CML-related deaths were considered. For the 60% of patients who remained on imatinib in the IRIS trial at last follow-up, nearly all had achieved at least a complete cytogenetic response (CCyR) and the progression rate in the years beyond the first 3 was close to zero.1 So far, no TKI has achieved superior survival compared with imatinib, although there may be an emerging trend for better progression-free survival with nilotinib and dasatinib.
One of the issues with imatinib that has not been clearly resolved is the optimal dose. Randomized studies comparing 400 and 800 mg/d did not demonstrate a clear advantage with the higher dose, except in the case of the German CML IV study, in which higher rates of molecular response (MR) were achieved with 800 mg/d. However, no evidence of an improvement in progression-free or overall survival has been observed in any of the randomized trials comparing different dosing schedules.
Nilotinib
Worldwide experience with nilotinib has increased greatly over the past 5 years. Although structurally similar to imatinib, its affinity for BCR-ABL and off-target effects are quite different. Nilotinib is a highly potent TKI that binds the kinase domain with greater affinity and is less vulnerable to kinase domain mutations. Only 5 mutations are of major concern (T315I, F359V, E255K/V, and Y253H). They are the mutations that most frequently emerge on frontline or second-line nilotinib therapy and their presence is a contraindication to the use of nilotinib after failure of another TKI.10,13
The day-to-day toxicity of nilotinib is generally quite favorable; edema is rare and gastrointestinal toxicity is uncommon. Elevated lipase and abnormal liver function tests are each observed in 5% to 10% of patients, but do not often lead to discontinuation of therapy.12 The ENESTnd study demonstrated that nilotinib resulted in superior rates of cytogenetic and MR and a lower rate of progression compared with imatinib at a dose of 400 mg/d. After a minimum follow-up of 4 years, major MR (MMR) and MR4.5 rates were higher in both the nilotinib 300 and 400 mg twice daily arms compared with the imatinib arm. Estimated 4-year rates of freedom from progression to the accelerated phase (AP) or blast crisis (BC) on study (including events during follow-up after discontinuation) were 97% (P = .05) and 98% (P = .0074) in the 300 and 400 mg twice daily nilotinib arms, respectively, compared with 93% on the imatinib arm. However, after 4 years, the lower risk of transformation had not translated into a survival advantage when all patients are considered.14 A critically important question that has not yet been addressed in published reports of ENESTnd is whether certain risk groups (eg, patients with high Sokal or EUTOS scores) may have superior survival if they receive nilotinib rather than imatinib.
An emerging concern related to the use of nilotinib is the occurrence of vascular events including peripheral arterial occlusive disease, coronary artery disease, and cerebrovascular disease, as well as hyperglycemia and hypercholesterolemia. Sporadic reports from single centers of cases with serious progressive vascular events emerged first, but most of these patients had multiple risk factors for vascular disease.15-17 We now have evidence from ENESTnd that the incidence of these 3 types of vascular events appears to be higher on both nilotinib arms than on the imatinib arm. A potential confounding factor is the possibility that imatinib actually reduces the probability of these vascular events. A recent analysis compared CML patients from 3 trials (IRIS, TOPS, ENESTnd) with patients who received non-TKI therapy (IFN plus Ara-C).18 Although rates of vascular events were higher in the nilotinib group compared with the imatinib group, there was no significant difference between the non-TKI group and the nilotinib group. How significant this problem will be with long-term exposure to nilotinib remains to be determined, but it is certainly an issue to keep in mind when selecting a TKI for a patient with risk factors for atherosclerotic disease and vascular occlusions. If nilotinib is chosen as frontline therapy, then careful attention to glucose and lipid levels is warranted. Furthermore, any clinically significant vascular event occurring on nilotinib therapy should signal the need to review the choice of ongoing TKI therapy.
Nilotinib is also associated with hyperglycemia, possibly by inducing insulin resistance. In the ENESTnd trial, 20% of patients on the nilotinib 300 mg arm who were not diabetic at baseline were diabetic by 3 years compared with 9% on the imatinib arm. Diabetic patients who are starting nilotinib therapy should be closely monitored.19
Dasatinib
Many CML patients have been treated with dasatinib in recent years. In vitro, dasatinib is the most potent of the 3 available TKIs, but in the clinical setting, it would appear to have similar potency to nilotinib (Table 1) when different drug scheduling and dosing is considered. A unique aspect of the pharmacokinetics of dasatinib is its relatively short half-life, suggesting that it would be optimally delivered in 2 or 3 divided daily doses. A well-designed randomized clinical trial (START-R) demonstrated that twice daily dosing offers no advantage in terms of efficacy or toxicity over once daily dosing. The frontline randomized trial DASISION trial comparing 100 mg daily of dasatinib to imatinib 400 mg daily demonstrated that dasatinib is generally well tolerated and achieves superior early MRs compared with imatinib. The rate of progression was lower on the dasatinib arm, but no survival advantage has emerged thus far.

Nil(300) indicates nilotinib 300 mg; IM, imatinib; DAS, dasatinib; and BOS, bosutinib.
*Median follow-up was 36 months.
†Median follow-up was 24 months.
‡Median follow-up was 18 months.
With regard to kinase domain mutations, there have been similar numbers of mutations on imatinib and dasatinib in the DASISION trial. A higher number of T315I mutations have been observed in this trial on the dasatinib arm, which is in contrast to the ENESTnd trial, where T315I was seen in a similar number of patients in all 3 arms. The reasons for this difference are not clear but may relate to the limited number of mutations that are resistant to dasatinib (T315I/A, V299L, F317L/I).11
Only 10% of patients have withdrawn from the dasatinib arm of the DASISION study because of adverse events,20 the major concern relating to pleural effusions. Approximately 20% to 25% of patients will develop pleural effusions, usually grade I-II, but frequently leading to the use of diuretics and steroids and the need to interrupt and/or reduce the dasatinib dose. Of more concern are rare reports of pulmonary arterial hypertension, which is a life-threatening condition. Nine cases were reported to the French pulmonary hypertension registry over a 4-year period.21 All but one of these cases improved markedly with dasatinib cessation, but none returned to normal pulmonary pressures and 2 of these patients have since died. The incidence was estimated to be 0.45% in French patients receiving dasatinib. The extent of the problem with long-term exposure will need to be closely monitored.
Intriguingly, clonal lymphocytosis with large granular lymphocyte morphology is not uncommon in dasatinib-treated patients and is associated with superior response rates. A Japanese group reported a ≥1.5-fold increase in the lymphocyte count associated with higher rates of complete MR (CMR) at 12 months (67% vs 26%, P = .0011) in patients receiving dasatinib for imatinib resistance.22 Whether this represents an immune response induced by dasatinib is unclear and further work is needed to ascertain whether this phenomenon could be exploited to further improve responses.
In summary, we have the original TKI imatinib, which is extremely safe and potentially quite cheap, but only leads to long-term optimal response in approximately 60% of patients, and we have the much more potent second-generation TKIs nilotinib and dasatinib, which probably reduce the transformation risk but do not appear to improve overall survival. They both have some question marks regarding long-term toxicity, which should lead us to be cautious about their widespread use without clear justification.
Deciding on the goals of therapy for your patient
Before deciding on the best therapeutic approach for a newly diagnosed CP-CML patient, it is important to be clear about the goals of therapy in each case (Figure 1). For elderly patients with substantial comorbidities, the main focus of CML therapy is likely to be prolonging survival by reducing the risk of progression. For the frail elderly patient who tolerates TKI therapy poorly, maintaining reasonable quality of life may become a higher priority than prolonging survival. For younger patients, maximizing the prospects of survival remains the main goal of therapy, but improving the prospect of treatment-free remission should now be considered as an additional goal. It is alarming to contemplate that a 15-year-old with CML might need to take a TKI continuously for disease control for the next 70 years unless treatment-free remission can be achieved. Although imatinib appears to be a safe drug over the course of 10 to 15 years of exposure, significant organ toxicities may be revealed with lifelong exposure. Young women who wish to start a family would also value the achievement of treatment-free remission very highly.

Proposed schema for individualizing therapy based on comorbidities, goals of therapy, and disease risk profile. For aggressive triggers, there should be a switch to more potent TKI (>10% at 3 months or >0.1% at 12 months).
Proposed schema for individualizing therapy based on comorbidities, goals of therapy, and disease risk profile. For aggressive triggers, there should be a switch to more potent TKI (>10% at 3 months or >0.1% at 12 months).
Is it reasonable and realistic to be considering treatment-free remission as a high priority goal of therapy based on the limited data we have available today? The evidence from the STIM trial and the Australian TWISTER trial is fairly convincing: ∼30% to 40% of CML patients who achieve a stable deep MR on imatinib can stop therapy and remain PCR negative for many years.23-25 In fact, there has been no evidence of late molecular recurrence in any of the patients who remained PCR negative for the first 27 months after imatinib cessation. Several hundred CML patients have now entered cessation studies and no reports of progression or drug resistance have emerged to date. We need to be cautious and realistic about considering treatment-free remission as a goal for younger patients, because only a small minority of patients who receive frontline imatinib will ever achieve it. We calculated in an Australian population of >400 CML patients that ∼35% will eventually achieve stable undetectable minimal residual disease (previously called stable CMR, denoting 2 years of monitoring in which every test demonstrated an absence of BCR-ABL by quantitative RT-PCR with sensitivity of at least 4.5 logs as determined by the number of control gene transcripts amplified).26 Of these patients, ∼30% are likely to remain in stable MR long term. Therefore, in a population of CML patients who receive frontline imatinib, <15% will eventually achieve treatment-free remission. Will we be able to achieve a substantially higher rate if we use the more potent TKIs frontline or second-line? This is a question under active investigation; however, we will not know the definitive answer for another 3+ years. It is a crucial question because the potential to improve the rate of treatment-free remission may be the strongest argument in favor of using frontline nilotinib or dasatinib in younger patients regardless of risk profile.
A recent French study assessing the molecular disease recurrence rate in patients ceasing second-generation TKI therapy after achieving stable deep MRs was encouraging. Patients who switched to nilotinib or dasatinib because of intolerance to imatinib and then achieved stable CMR had a 60% probability of remaining in remission off therapy.27 Given that the achievement of deep MRs appears to be higher with second-generation TKIs than with imatinib, the overall rate of treatment-free remission achieved using nilotinib or dasatinib frontline may be significantly higher than the 15% who can achieve it on imatinib (Table 2). On this basis, we can conclude that, on the balance of probabilities, it is likely that second-generation drugs used frontline will achieve a higher rate of treatment-free remission overall. However, more mature data are needed before we can say that the case is proven beyond reasonable doubt.
Actual rates of stable undetectable minimal residual disease and sustained remission after cessation and calculated rates of overall treatment-free remission for imatinib and estimated rates for second-generation TKIs
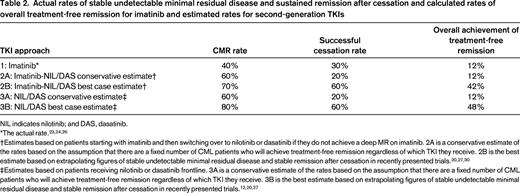
NIL indicates nilotinib; and DAS, dasatinib.
†Estimates based on patients starting with imatinib and then switching over to nilotinib or dasatinib if they do not achieve a deep MR on imatinib. 2A is a conservative estimate of the rates based on the assumption that there are a fixed number of CML patients who will achieve treatment-free remission regardless of which TKI they receive. 2B is the best estimate based on extrapolating figures of stable undetectable minimal residual disease and stable remission after cessation in recently presented trials.20,27,30
‡Estimates based on patients receiving nilotinib or dasatinib frontline. 3A is a conservative estimate of the rates based on the assumption that there are a fixed number of CML patients who will achieve treatment-free remission regardless of which TKI they receive. 3B is the best estimate based on extrapolating figures of stable undetectable minimal residual disease and stable remission after cessation in recently presented trials.12,20,27
Achieving the goals of therapy
Survival only
The ENESTnd, DASISION, and ECOG studies were all powered to identify a superior rate of cytogenetic or molecular response for the investigational TKI at 12 months.12,14,28 Although all of these studies have shown superior response rates for the more potent TKIs compared with imatinib, none has demonstrated a survival advantage over imatinib in the frontline setting so far. Therefore, when considering frontline therapy in the older patient, in whom survival is the predominant goal, imatinib remains an excellent choice. If imatinib is the frontline choice, then it is still critical (perhaps even more critical) that cytogenetic and molecular response are closely monitored. Under the European LeukemiaNet (ELN) recommendations, criteria for failure represent an indication to switch therapy because survival is likely to be inferior if current therapy is maintained. The molecular targets in this circumstance should be BCR-ABL values measured by quantitative RT-PCR on the international scale of <10% and 1% by 6 and 12 months, respectively. A BCR-ABL level <0.1% (MMR) by 18 months is also optimal; however, the additional benefit of achieving MMR by 18 months, compared with achieving CCyR or its molecular equivalent, <1% BCR-ABL, in terms of the long-term prospects of survival is only small. There is a 1% versus 3% risk of death over the subsequent 5 years for patients who, at 18 months, achieved ≤0.1% and those patients between 1% and 0.1%, respectively.29 Should these goals be any different for high-risk patients in whom the sole focus is survival? At this stage, we do not have any evidence that molecular and cytogenetic targets should be any different for high-risk patients.
Survival and treatment-free remission
For patients with a (non-CML-related) life expectancy of more than 10 years, the achievement of a deep MR will usually be a high priority because it brings with it the possibility of a trial of drug cessation and treatment-free remission. Assuming that the clinician and patient accept treatment-free remission as a long-term goal, how does this change the molecular targets of therapy? A study of >400 patients in Australia demonstrated that the key molecular targets that were predictive for the achievement of stable undetectable MRD on ongoing imatinib therapy were a BCR-ABL value <10% by 3 months and <0.1% by 12 months.26 The more potent TKIs have a clear advantage over imatinib in achieving these molecular landmarks. This conclusion is supported by the higher rates of deep MRs observed at 2 years and beyond with nilotinib or dasatinib in the phase 3 trials compared with imatinib (Table 1).
Relying on the early MR to identify the high-risk patient
One strategy designed to maximize the use of imatinib and only use more potent TKIs where there is evidence of a high risk of progression is to use frontline imatinib and rely on the MR at 3 and/or 6 months to identify the high-risk patients and switch them to a more potent TKI. This was the rationale for the TIDEL II study in Australia. Patients who were >10% BCR-ABL at 3 months were dose escalated to imatinib 800 mg and, if still >10% at 6 months, switched to nilotinib (cohort 1, n = 105) or switched at 3 months straight to nilotinib if they were >10% (cohort 2, n = 105). The final analysis is not yet complete, but it is clear that this approach does not rescue all patients from an adverse outcome. The rate of transformation for patients who were >10% BCR-ABL at 3 months was >10% and progression events among these high-risk patients were usually observed in the first 12 months.30 We can only speculate as to whether these patients would have achieved better outcomes if they had started nilotinib as frontline therapy. The ENESTnd study demonstrated adverse outcomes for patients >10% at 3 months whether they received frontline nilotinib or imatinib. The risk of transformation was >10% in both arms and half of the progression events occurred before 6 months. These observations suggest that, although the 3-month MR is a good indicator of the long-term probability of achieving a deep MR and the short-term risk of progression, it has limited value as an indicator of high risk for the purposes of intensifying therapy. This is because 3 months will often be too late to reverse an adverse outcome.
Risk-adapted therapy
Another important consideration when choosing frontline therapy is the patient's prognostic score. There are 3 scoring systems that are currently being applied in CML, the Sokal, Hasford, and EUTOS systems, and there is no clear indication that one is superior to the others. Regardless of which scoring system is used, a high score is associated with a higher risk of progression to AP or BC. Because both nilotinib and dasatinib have been shown to reduce the risk of CML progression, these drugs might be preferred over imatinib in this group of high-risk patients.
There are many other predictive markers that may prove to be better than the current risk scores or, more likely, provide additional predictive value. However, these predictors are either not widely available, have not been prospectively established as independent predictors, or both. Current recommendations cannot incorporate these biomarkers, but in the next section we cover the most promising candidates.
Future prospects in optimizing frontline therapy
A better strategy might be to select the optimal TKI on the basis of biological markers of risk or treatment response. Patients with a high risk of progression or drug resistance could potentially be identified at diagnosis and treated aggressively on investigational protocols. To be valuable in this setting, the assay(s) must predict with a high degree of accuracy those patients destined to respond poorly to imatinib. In addition, the assay should ideally describe the underlying resistance mechanism such that the next TKI or therapeutic strategy can be rationally selected. Although there have been many assays described, it is not clear whether any will tick all of these boxes.
IC50 analysis
In 2005, we described an in vitro assay, based on phosphorylation of the adaptor protein Crkl to determine the response to imatinib in patient's CML cells collected at diagnosis. This study demonstrated substantial interpatient variability in sensitivity. Furthermore, patients whose cells were more sensitive to imatinib (greater decrease in p-Crkl) kinase inhibition (low IC50) achieved 1% BCR-ABL by 3 months and 0.1% by 12 months at a significantly higher rate than patients with cells less sensitive to kinase inhibition (high IC50; P = .01 and P = .03, respectively.3 That study demonstrated that the degree of kinase inhibition achieved was a critical determinant of subsequent MR and that patient-specific factors at the time of diagnosis determine this sensitivity. This assay can also be performed for other TKIs, but to date there are no published clinical correlations between the IC50 results for nilotinib or dasatinib and subsequent MR. It therefore remains unclear whether this assay can be used to individualize frontline patient therapy aside from predicting which patients are destined to perform poorly on imatinib.
OCT-1 activity
OCT-1 is the major active influx pump for imatinib into target leukemic cells.6,7 OCT-1 activity performed on diagnostic blood cells before the initiation of therapy is a strong predictor of subsequent MR at both 24 and 60 months, as well as predicting progression and event-free survival in imatinib-treated patients.8,9,31 There is also some evidence from the TIDEL II study that patients with low OCT-1 activity who fail to achieve early, time-dependent molecular targets also have suboptimal responses when switched to nilotinib.32 This suggests that OCT-1 activity may also provide a surrogate marker of underlying disease biology and those patients with disease characterized by low OCT-1 activity may respond poorly to all TKIs. However, only frontline trials of nilotinib and dasatinib will provide direct evidence to test this hypothesis, and these data are not currently available. Regardless, there is now good evidence to suggest that the majority of patients with high OCT-1 activity will achieve excellent responses on imatinib (60 months: transformation-free survival 100%, overall survival 96%, P = .002 and P = .02, respectively, compared with patients with low OCT-1 activity).33 Conversely, patients with low OCT-1 activity are at risk of poor response to imatinib and require proactive monitoring and intervention.
CIP2A
It has been demonstrated previously that high expression levels of the cancerous inhibitor of PP2A (CIP2A) are associated with aggressive forms of both breast and gastric cancers. Recently, Lucas et al demonstrated in a cohort of 31 CML patients that CIP2A protein levels were significantly higher in the diagnostic cells of patients who subsequently transformed (P < .001).34 This was associated with higher levels of both PP2A protein and phosphorylation and increased levels of c-Myc. Although this result awaits validation in an independent cohort, the finding that CIP2A is involved in progression is important and reveals CIP2A as a potential therapeutic target.
GFI-1
Soliera et al recently demonstrated that ectopic GFI1 expression inhibited proliferation and colony formation both in p210BCR/ABL–expressing cell lines and in primary CD34+ CML cells through the repression of STAT5B and/or Mcl-1.35 That study, along with their previous work,36 demonstrated the biological importance of the GFI1/STAT5B/Mcl-1 regulatory pathway on proliferation and survival of CML cells. In a study of CP-CML patients, Kok et al demonstrated that decreased GFI1 expression at diagnosis is highly correlative with disease progression and transformation to BC.37 These results suggest that patients who have low GFI1 expression have a high risk of early transformation, supporting the previously described role of GFI1 in the inhibition of proliferation and colony formation of p210BCR/ABL–transformed cells and primary CD34+ CML cells and supporting GFI1 as a candidate biomarker for high-risk CML.
Has the “omics” era improved prognostication in CML?
The variable responses of patients to imatinib therapy suggests that CP-CML is not a homogeneous disease and that underlying biology intrinsic to the patient's leukemia plays a key role in response determination. This finding has led to several studies of gene expression profiling in which patients were grouped as achieving or not achieving various response milestones. In a hallmark analysis, Radich et al identified gene expression changes associated with progression.38 McWheeney et al demonstrated in CD34+ cells that the ontogeny of the gene set identified in patients failing to respond to imatinib resembled that of advanced disease as identified by others.39 Although there have been many such studies performed in the imatinib era, each demonstrating compelling gene sets that appear to provide predictive value and some biological insights, there is disappointingly little overlap in the genes identified by these studies. This is most likely related to the differing criteria used to subdivide the patient groups, the differing cell types (mononuclear cells vs CD34+ cells), and possibly also the platforms used. To date, despite the number of studies performed, gene expression profiling has not advanced the biomarker area significantly. There have, however, been several candidate gene approaches in recent years that have identified biomarkers.
BIM polymorphism
Recently, Ng et al identified a common 2903 bp deletion in the noncoding region of the BIM gene that is linked to the BCL2 family of cell death genes.40 This deletion results in the removal of the cell death activation domain of BIM and, importantly, provides a useful biomarker to predict those patients at risk for imatinib resistance. These researchers demonstrated that more than two-thirds of patients with the BIM deletion are resistant to imatinib therapy. The caveat here is that this gene deletion is rarely found outside of Asian populations, so whereas screening for this BIM deletion may be clinically helpful in Asian cohorts, it is unlikely to be of value outside of this population. However, this study provided critical evidence for the importance of the BCL2 pathway in mediating TKI-related cell death, paving the way to search for gene deletions or polymorphisms in other ethnicities.
Although there are currently no biomarkers that can clearly define the “optimal” therapy for each patient at the time of diagnosis, it is likely when the data from upfront nilotinib and dasatinib studies matures, such biomarkers may be identified. The overriding problem with biomarker development is that outcome measures can be muddied by issues of compliance and tolerance. It is important to know whether a patient fails to respond because they have unfavorable disease biology or because they are poorly adherent with their TKI therapy. One important aspect that has been revealed in the quest for potent biomarkers is that various underlying biologies can be targeted therapeutically. BIM deletions could potentially be overcome by using BH3 mimetics,40 OCT-1 activity can be enhanced in vitro using diclofenac,41 and CIP2A and GFI1 may provide a further drug target to overcome transformation events. However, whereas such approaches have appeal in vitro, all require in vivo validation, most likely in the setting of a combination approach with TKI therapy. As our understanding of underlying disease biology grows, combinational strategies designed to inhibit both tyrosine kinase and other targets may become the focus of investigational protocols of the future.
Second-line therapy
One of the justifications used for selecting imatinib as frontline therapy is that it leaves a full range of more potent TKIs for second-line therapy. There is a general consensus that patients who fail imatinib therapy (see ELN guidelines for failure) should switch without hesitation to either nilotinib or dasatinib. The choice would be guided by the mutation profile, if relevant, and the comorbidities of the patient. The probability of achieving a good response to second-line therapy can be predicted based on a few baseline variables.42,43 One of the strongest predictors of response to second-line therapy is response to first-line therapy. In an MD Anderson study, 3-year event-free survival for patients who had achieved MCyR on imatinib before treatment failure was 67% after switching to second-line therapy compared with 30% in patients with no prior cytogenetic response to imatinib. Furthermore, the success of switching can be assessed quite early based on the MR at 3 months.44 For patients who fail nilotinib or dasatinib frontline therapy, the course of action is less clear. The salvage rate when switching from dasatinib to nilotinib or vice versa is not well documented. In the case of mutations that are clearly susceptible to the second-line drug, this may be an effective approach, but for most patients, it is unlikely to be effective in the long term. The PACE trial demonstrated that in the second-line and even in the third-line setting, there was a reasonable expectation of response to ponatinib, even in patients who had already failed 2 or more TKIs.45
Other TKIs that may be frontline soon
Bosutinib
Bosutinib is a potent second-generation TKI that also has SRC inhibitory activity. In the BELA phase 3 randomized study, it failed to demonstrate superior rates of CCyR compared with imatinib at 12 months.46 However, when the investigational arms of the 3 randomized studies are put alongside each other, the response and progression rates were similar to those achieved with nilotinib and dasatinib (Table 1). Further studies may establish a place for bosutinib in the frontline setting. For the moment, bosutinib is registered in many countries as a second- or third-line option. In a phase 2 study, 23% of patients who were resistant to imatinib and either nilotinib or dasatinib achieved CCyR on bosutinib. Discouragingly, only 24% are still on bosutinib in this study.47
Ponatinib
There has been a lot of excitement about the only clinically available TKI that has activity against the T315I mutation. This mutation is resistant to all other currently available TKIs, so for patients with this mutation, outside of the allograft option, ponatinib represents the only opportunity to achieve long-term disease control. However, its activity is not confined to the T315I mutant form of BCR-ABL. The phase 1 and PACE studies have demonstrated that a high proportion of patients resistant to 2 or more TKIs will achieve excellent responses to ponatinib. Even though patients with the T315I mutation had higher rates of response (MCyR rates of 70% in T315I patients vs 50% in other resistant/intolerant patients, median follow-up 11 months),45 a multivariate analysis found that younger age and dose intensity were the significant predictors of response, not the presence of the T315I mutation.48 A potential drawback is the toxicity profile. Pancreatitis can be a significant problem and an association with vascular events that has recently been recognized requires further clarification.
Other potential frontline approaches
Combination pegylated IFN plus a TKI
Alpha IFN was the best drug available for CML before the TKI era. Although monotherapy with alpha IFN is rarely indicated now in any circumstance except pregnancy, it has a potential role in combination with a TKI. Although the German CML IV study did not demonstrate an advantage for patients who were given imatinib plus alpha IFN, 2 other randomized studies show significantly better responses in patients given pegylated IFN with imatinib. Both the SPIRIT and the Nordic study showed that relatively low doses of pegylated IFN given for several months in the first year was associated with superior rates of deep MR.49 No reduction in the probability of progression or death has been observed to date. This remains an experimental approach, but may be a promising strategy to achieve deeper MRs and potentially recruit more patients to cessation studies.
Allograft: when is it the right option?
With the recent availability of ponatinib, there has been a need to modify the indications for allogeneic transplantation in CML. Previously, patients who developed the T315I mutation were considered for an allograft because none of the available TKIs had any activity against this mutation. Now, ponatinib may be a reasonable choice for these patients. So who should receive an allograft for CML in 2013? There are 3 possible categories: (1) any patient who presents in BC or progresses to AP or BC on therapy should be considered for an allograft as soon as chronic phase has been reestablished; (2) patients who fail ponatinib therapy after failing a second-generation TKI are very unlikely to respond to another TKI; and (3) children with CML who fail to achieve MCyR or a BCR-ABL level <10% by 6 months despite receiving dasatinib or nilotinib therapy. Although ponatinib could be used in this setting, its safety data are limited at present and the potential for emerging toxicity must be considered when choosing treatment for a child.
Conclusions
For many physicians, there is an embarrassment of riches when it comes to therapeutic options in CML. When choosing frontline TKI treatment, the clinician must consider what is known about CML biology and the safety and efficacy profiles of the 3 TKIs to achieve the best possible outcome for the patient. Key issues to consider are the patient's long-term goals, the intrinsic risk profile of the leukemia, and patient comorbidities. The emerging importance of treatment-free remission as a goal of therapy will likely assume increasing prominence in the treatment paradigm. The recent availability of ponatinib has expanded our second- and third-line options and further reduced the pool of patients who will need to be considered for allografts. Recent progress in the development of predictive assays that can accurately identify high-risk patients should provide a solid basis for the development of customized therapy over the next decade.
Disclosures
Conflict-of-interest disclosure: T.H. is on the board of directors or an advisory committee for, received research funding from, and received honoraria from Novartis, BMS, and Ariad and has been affiliated with the speakers' bureau for Novartis. D.W. has received research funding from BMS and Novartis and has received honoraria from BMS and Novartis. Off-label drug use: None disclosed.
Correspondence
Timothy Hughes, Department of Haematology, SA Pathology, Frome Road, Adelaide 5000, SA, Australia; Phone: +618 82223330; Fax: +618 82223139; e-mail: timothy.hughes@health.sa.gov.au.
