
Abstract
In patients with chronic myeloid leukemia (CML) in chronic phase who have achieved complete molecular remission on imatinib therapy, clinical trials from France and Australia have demonstrated that the majority experience prompt molecular relapse of their leukemia upon discontinuation of the drug, showing that long-term monotherapy with tyrosine kinase inhibitors is not curative in the majority of patients with CML. This has focused attention on strategies to eradicate residual disease in CML that is presumed to arise from malignant Ph+ stem cells, which should result in permanent cure and long-term leukemia-free survival. Here, we review the evidence that targeting CML stem cells will be of clinical benefit and discuss pharmacological and immunological approaches to accomplish this goal. Where possible, we link preclinical studies of CML stem cell biology to emerging results from clinical trials of agents that may target these cells.
Introduction
A primary clinical goal of all oncologists is to cure patients of their malignant disease. For most patients with chronic myeloid leukemia (CML), the advent of ABL1 tyrosine kinase inhibitor (TKI) therapy has turned their chronic leukemia into a truly indolent medical condition, the management of which bears more resemblance to diabetes or hypertension than to cancer. Epidemiological studies have shown that the relative survival of CML patients treated since the widespread introduction of imatinib in 2001 is approaching 90% of age-matched controls,1 whereas the survival of CML patients who achieve complete cytogenetic remission (CCyR) within 2 years of starting imatinib is not statistically different from the general population.2 This has led to the concept of “operational” or “functional” cure of CML,3 and begs the question of whether the goal of achieving a TKI- and leukemia-free state is necessary or even desirable for CML patients.4
We would argue in the affirmative. Although TKI therapy is generally well tolerated in patients with CML, we do not understand fully the influence of chronic, long-term grade 1 to 2 toxicity on quality of life and on treatment adherence, which has a major impact on clinical outcome,5 nor do we know whether lifelong TKI therapy will have untoward adverse effects that are manifest only after decades of treatment. Women of childbearing age who wish to conceive should probably discontinue TKI treatment during pregnancy and nursing.6 The burden of the cost of TKI therapy on both CML patients and society has become an issue,7 one that will increase in importance as the prevalence of CML rises.8 Lastly, it is plausible that treatment strategies found to eliminate CML stem cells in patients in cytogenetic or molecular remission might be extended to those patients who have a suboptimal early molecular response to TKI therapy, defined as a > 10% level of BCR-ABL1 transcripts at 3 months. Such patients have a decreased probability of achieving CCyR or major molecular response (MMR, defined as < 0.1% BCR-ABL1 transcripts) and increased risk of progression to accelerated phase or blast crisis,9,10 and this inferior prognosis might not be ameliorated by early switching to another TKI.11
Whither CML stem cells?
The cancer stem cell hypothesis states that cancers are heterogeneous and only a minor population of tumor cells has the properties, most rigorously assessed by transplantation, of self-renewal and tumorigenicity.12 Although this theory may not hold for certain solid tumors13 and even some hematologic malignancies,14 in CML, the evidence for leukemia stem cells (LSCs), while imperfect, is fairly convincing. It has long been appreciated that the Ph chromosome translocation in CML occurs in a pluripotent hematopoietic progenitor, whereas more recent studies demonstrate that this progenitor must have the stem cell–like capacity to self-renew because BCR-ABL1 expression does not confer self-renewal upon committed hematopoietic progenitors that lack this property.15 Xenotransplantation of primary CML cells into immunodeficient SCID mice was achieved in 1996 and provided the first functional evidence of CML stem cells,16 but the efficiency of Ph+ engraftment is low and highly variable even in more immunocompromised NOD/SCID or NOD/SCID/Il2rg−/− (NSG) recipients,17 making quantitation of these LSCs difficult. The immunophenotype (Lin−CD34+CD38−CD90+) of the CML cells capable of engrafting immunodeficient mice is concordant with that of normal human hematopoietic stem cells (HSCs), and BCR-ABL1+ cells with this immunophenotype are present in CML BM at frequencies similar to normal HSCs.18 Studies in mouse models of CML in which BCR-ABL1 is expressed via retroviral or transgenic methods in primary BM cells also demonstrate that the cells capable of transplanting CML-like leukemia to syngeneic recipients have the same immunophenotype as normal mouse HSC (Lin−Sca-1+c-Kit+; LSK).19,20 These observations suggest that CML stem cells do exist and are phenotypically and functionally quite similar to normal HSCs.
If we accept this working definition of CML stem cells, then several lines of evidence suggest that these cells are not effectively killed by TKIs in vitro nor are they efficiently eliminated from patients taking these drugs. The seminal studies of Holyoake et al demonstrated that Lin−CD34+ CML progenitors can remain quiescent in vitro in the presence of growth factors, can engraft NSG mice, and are insensitive to killing by imatinib,21 whereas Bhatia et al identified similar BCR-ABL1+ CD34+ progenitors in the BM of imatinib-treated CML patients in CCyR.22 Subsequent studies have shown that imatinib and more potent second-generation ABL1 TKIs are unable to eliminate these quiescent CML stem cells despite virtually complete inhibition of BCR-ABL1 kinase activity,23-25 suggesting that these LSCs are not “oncogene addicted,” (ie, they are not absolutely dependent on BCR-ABL1 for survival). Consistent with this, residual Ph+ progenitors isolated from CML patients in MMR or complete molecular response (CMR) seem to have lower BCR-ABL1 transcript levels than are found in similar progenitors from patients at diagnosis.26,27 After the initiation of TKI therapy, BCR-ABL1 transcripts measured in blood or BM decline logarithmically with several distinct phases or slopes.28,29 Although different mathematic models can be derived from these data, a consensus interpretation is that the initial rapid decline in transcripts over the first ∼ 6 months of treatment represents elimination of differentiated leukemic cells, with slower phases over subsequent years reflecting depletion of immature progenitors and perhaps LSCs.30,31
Functional or operational cure of CML is defined by sustained molecular remission upon cessation of TKI treatment. Although there are many anecdotal reports of outcomes after imatinib discontinuation, our best insights come from clinical trials of TKI cessation in CML patients who were in CMR for at least 2 years: the STIM trial from France32 and the CML8 trial from the Australian Leukaemia and Lymphoma Group.33 Both studies yielded similar results with molecular relapse rates of ∼ 60%, the majority of which occurred in the first 6 months. High Sokal risk at presentation was the only predictor of relapse risk and resumption of imatinib treatment resulted in restoration of molecular remission in the majority of patients. Interestingly, the use of DNA-based BCR-ABL1 PCR, which is 1 to 2 logs more sensitive that RT-PCR, showed detectable BCR-ABL1 fusion genes in most patients who sustained CMR off of TKI therapy.34 A preliminary report of cessation of therapy in 42 CML patients with CMR induced by the more potent second-generation TKIs (dasatinib and nilotinib) found that 58% experienced a molecular relapse to > 0.1% BCR-ABL1 transcripts (ie, loss of MMR), whereas 7 (17%) remained in CMR and 11 (26%) exhibited low-level but detectable BCR-ABL1 transcripts on at least one occasion.35 The long-term outcome for patients with detectable BCR-ABL1+ cells who do not resume TKI treatment is unknown, but the situation may be analogous to allografted CML patients, who may also exhibit low-level fluctuations in BCR-ABL1 transcripts without clinical relapse.36
Collectively, what do these translational and clinical studies tell us? First, it is plausible that a small fraction of the patients who are in treatment-free CMR after TKI cessation are in fact cured of their disease, because some patients have now been off therapy for more than 4 years. Although late relapses (> 5 years after transplantation) can occur in allografted CML patients who achieve stable CMR,37 these are rare and long-term treatment-free remissions have also been described in CML patients treated only with IFN-α.38,39 Second, it is clear that eradication of all detectable Ph+ cells is not a prerequisite for long-term disease-free survival in CML regardless of the treatment approach (allografting, IFN-α, or TKIs), implying that low levels of LSCs can be held in check by physiological or immunological mechanisms. Third, although it has not yet been tested in the clinical setting, it is likely that the relapse rate after TKI cessation in CML patients who are not in CMR will be much higher than 60% and may approach 100%. Fourth, for CML patients not in CMR and for the majority of those who achieve CMR on TKI therapy, additional treatment approaches will be needed to eliminate residual LSCs that contribute to relapse. The remainder of this article will focus on these strategies (Table 1).40,41
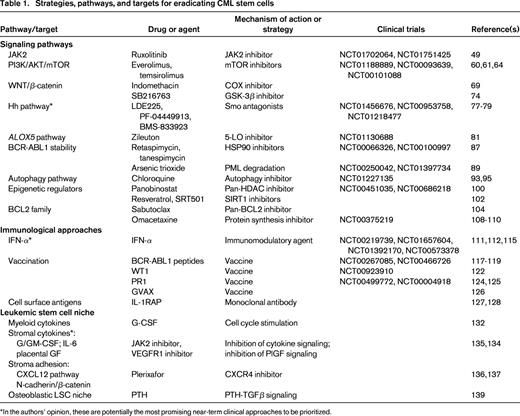
Targeting signaling pathways implicated in CML stem cell survival
Although normal HSCs and Ph+ LSCs express similar cell surface markers, they differ significantly in signaling. BCR-ABL1 activates a myriad of signaling pathways in hematopoietic cells, some of which are shared with those affecting normal HSCs.42 Not all of these pathways appear to be required for leukemogenesis and it is likely that only a subset will contribute to the maintenance of the most primitive CML stem cells (Figure 1). How does one identify these signaling pathways, which could be mined for therapeutic targets? Whereas CML stem cells can be defined and quantified in part by their immunophenotype, but most rigorously through transplantation, it follows that that studies aiming to define the physiology of CML stem cells must be carried out either using primary CML cells and surrogate in vitro (long-term culture-initiating cells [LTCICs]) or xenotransplantation assays or in physiologically accurate and quantitative mouse models of CML. The latter model systems have played a large role in this effort. If we accept the notion that complete or nearly complete inhibition of BCR-ABL1 by TKIs is insufficient to kill CML stem cells, it follows that the pathways of interest are either activated independently of BCR-ABL1 kinase activity or (perhaps more likely) are dependent on very low levels of this function. In either case, at least some degree of selectivity of targeting the pathway for LSCs versus normal HSCs will be necessary if the strategy is to be clinically relevant.

Signaling pathways and potential targets for elimination of the malignant clone in CML. Shown is schematic representation of the signaling pathways discussed that may contribute to the proliferation and survival of CML stem cells. Phosphorylation is represented by red “P” symbols. Arrowheads indicate activation of a downstream molecule. A bar at the end of a line indicates inhibition of a downstream molecule.
Signaling pathways and potential targets for elimination of the malignant clone in CML. Shown is schematic representation of the signaling pathways discussed that may contribute to the proliferation and survival of CML stem cells. Phosphorylation is represented by red “P” symbols. Arrowheads indicate activation of a downstream molecule. A bar at the end of a line indicates inhibition of a downstream molecule.
JAK-STAT pathway
Activation of the JAK-STAT pathway, principally involving JAK2 and STAT5, was first demonstrated in BCR-ABL1–expressing hematopoietic cells in 1996.43 Since that time, numerous studies have examined the contribution of JAK-STAT signaling to the pathogenesis of CML and, to a lesser extent, to the survival of CML stem cells. Despite this effort, the role of JAK kinases in CML pathogenesis is still not understood fully. Several JAK kinase family members, including JAK2, are activated in BCR-ABL1–expressing cells.43,44 JAK2 complexes with BCR-ABL1 via the ABL1 C-terminus and contributes to activation of the SRC kinase LYN,45 but neither the JAK2-binding site on BCR-ABL146 nor JAK2 itself47 is required for induction of CML-like leukemia in mice by BCR-ABL1. JAK2 kinase inhibitors decrease the proliferation and survival of cultured BCR-ABL1–expressing cells,48 but JAK2-deficient progenitors are equally sensitive to these drugs,47 indicating that targets other than JAK2 are responsible. However, these studies do not exclude a role for JAK2 in the maintenance of CML LSCs. A recent study demonstrated that the adapter protein AHI-1 mediates physical interaction between BCR-ABL1 and JAK2 in primitive CML progenitors, whereas combined treatment with imatinib and a JAK2 inhibitor (TG101209) increased apoptosis and decreased the leukemia-initiating activity of these progenitors in NSG recipients.49 In addition to inhibiting JAK2 in CML stem cells, there is also a potential rationale for targeting JAK2 in the BM microenvironment in CML (see “Targeting the CML stem cell niche” below). Given that the safety and efficacy of a potent JAK2 inhibitor (ruxolitinib) in myelofibrosis has been established, the hypothesis that JAK2 inhibition might contribute to elimination of CML stem cells can now be readily tested in the clinical setting. Phase 1 studies of ruxolitinib in combination with ABL1 TKIs in CML with residual disease (www.clinicaltrials.gov identifiers NCT01702064 and NCT01751425) are being launched.
The transcription factor STAT5 is a major substrate of JAK2, but whether inhibiting STAT5 would effectively target CML stem cells is controversial. Constitutively active mutants of STAT5a induce CML-like leukemia in primary mouse hematopoietic cells,50 whereas deletion of STAT5 abolishes fatal CML-like leukemia induced by BCR-ABL1.51,52 However, there is also some evidence that, in mice, both normal HSCs51,52 and their BCR-ABL1–expressing counterparts52 can survive and contribute to hematopoiesis and disease progression without STAT5. In contrast, studies in primary human CD34+ cells demonstrated that knock-down of STAT5B inhibits clonogenicity and LTCIC function of both normal and CML progenitors.53 Whereas small-molecule inhibitors of STAT5 have been described recently,54 the utility of STAT5 inhibition in CML could soon be tested directly in the clinical setting.
PI3K/AKT/mTOR pathway
The PI3K/AKT pathway is activated by the BCR-ABL1 kinase through the GAB2 adapter protein,55 and many downstream targets of the PI3K/AKT pathway contribute to BCR-ABL1–induced leukemogenesis. However, relatively little is known about the role of this complex pathway in the regulation of CML stem cells. In Ph+ myeloid progenitors, activated AKT phosphorylates the FOXO3a transcription factor, causing its retention in the cytoplasm. In contrast, the most primitive CML stem cells display inactive AKT, nuclear FOXO3a, and nuclear phospho-SMAD2/3, the latter a hallmark of TGF-β signaling.56 When CML was induced in mice from donor stem cells lacking FOXO3a, the phenotypic LSC population and efficiency of leukemia induction declined upon serial transplantation, the most rigorous test of LSC function. These data suggest that autocrine TGF-β signaling, through an unknown mechanism, may suppress AKT inhibition of FOXO3a in CML LSCs. This raises the question of whether TGF-β antagonists, such as inhibitors of TGF-β receptor kinase, might be of benefit in eliminating residual disease in CML. Another twist in the story emerged with the discovery that BCL6, a leucine zipper transcriptional repressor overexpressed in B-lymphoma, is another downstream target of FOXO3a in CML stem cells that is up-regulated by ABL1 TKIs and represses ARF and p53.57 Targeting BCL6 with a novel peptidomimetic inhibitor of BCL6 corepressor binding, RI-BPI,58 inhibited BCR-ABL1 leukemogenesis in mice and eradicated CD34+CD38− CML progenitors in vitro.57 However, it is not clear whether this compound will move forward in the clinical setting.
The serine/threonine kinase mammalian target of rapamycin (mTOR) is a downstream target of PI3K/AKT that regulates mRNA translation in mammalian cells, controlling cell growth and proliferation. The mTOR protein functions as the catalytic subunit for 2 distinct protein kinase complexes, mTORC1 and mTORC2, which play important roles in growth and survival of BCR-ABL1–transformed cells.59 The combination of the mTORC1 inhibitor rapamycin and imatinib prolonged survival in the retroviral CML model and was effective against disease induced by imatinib-resistant mutants of BCR-ABL1.59 More recently, ATP-competitive dual mTORC1/2 inhibitors have exhibited potent growth inhibition in BCR-ABL1+ cell lines and primary CML cells.60,61 Indirect suppression of the mTOR function can also be achieved by modulating the AMP-activated protein kinase (AMPK) pathway.62 Stimulation of AMPK by resveratrol inhibits mTOR and has antileukemic effects in both imatinib-sensitive and imatinib-resistant CML cells.63 However, a recent study suggests that the clinical effects of mTOR inhibition in CML might be limited to more differentiated cells rather than LSCs.64 Two allosteric “rapalog” TORC1 inhibitors (everolimus and temsirolimus) are Food and Drug Administration (FDA)–approved in renal cancer, whereas multiple direct TORC1/2 inhibitors (OSI-027, AZD8055, MLN0128, CC-223) are in early-phase clinical trials in cancer. Several phase 1 trials of rapalogs in combination with ABL1 TKIs in CML are in progress (www.clinicaltrials.gov identifiers NCT01188889, NCT00093639, and NCT00101088).
WNT/β-catenin pathway
Abnormal WNT/β-catenin signaling was first linked to CML by the discovery of aberrant constitutive nuclear β-catenin in granulocyte-macrophage progenitors in patients with CML myeloid blast crisis (mBC).18 Whereas nuclear β-catenin expression is normally restricted to the HSC compartment, its misexpression was hypothesized to contribute to abnormal self-renewal in malignant granulocyte-macrophage progenitors, which comprise the LSCs in CML-mBC. Consistent with this, gene expression analysis showed increased expression of several WNT target genes in accelerated phase and mBC CML.65 The mechanism of β-catenin overexpression in CML-mBC cells is not clear, but aberrant splicing of transcripts for GSK-3β, a serine-threonine kinase that negatively regulates β-catenin through phosphorylation and subsequent proteasomal degradation, has been identified in these progenitors.66 Subsequent studies in the mouse retroviral model of CML demonstrated that deletion of β-catenin impairs the development of CML-like leukemia induced by BCR-ABL1.67,68 A subsequent study used conditional deletion to demonstrate that β-catenin is essential for the maintenance of CML stem cells in the retroviral model,69 whereas previous studies indicated that β-catenin is not required for survival of normal HSCs in the adult.70 Therefore, targeting β-catenin may be a novel strategy for selective elimination of CML stem cells. As yet, there are no agents in clinical trials that directly antagonize WNT signaling, but some small-molecule WNT pathway inhibitors have been shown to reduce β-catenin and induce apoptosis in primary CML cells.71,72 Cyclooxygenase inhibitors such as indomethacin appear to destabilize β-catenin by inhibiting prostaglandin E2 synthesis,73 and indomethacin treatment of mice with BCR-ABL1–induced CML lowered β-catenin levels and reduced CML LSCs as assessed by secondary transplantation.69 Phosphorylation of the eukaryotic translation initiation factor eIF4E by MAPK-interacting kinase (MNK) activates β-catenin via AKT in CML-mBC LSCs, and a MNK inhibitor (CGP57380) abolished long-term engraftment of CML-mBC LSCs in NSG mice.140 Interestingly, although GSK-3β negatively regulates β-catenin, treatment with an inhibitor of GSK-3β (SB216763) in combination with imatinib was found to decrease CML stem cells in vitro (as assessed by leukemic LTCIC), but this might be mediated by effects on other substrates such as p27.74
Hedgehog pathway
Hedgehog (Hh) signaling controls the response to stress, injury, healing, and regeneration and plays a critical role in the self-renewal of somatic stem cells. Binding of Hh ligands to their receptor, Patched (PTCH), results in the activation of Smoothened (SMO), which promotes the nuclear translocation of the GLI family of transcription factors (GLI1-3). The GLI family modulates the expression of genes such as Cyclin D, c-MYC, and BCL2 and thus controls cell proliferation and survival. Several lines of evidence implicate the Hh pathway in CML (for review, see Jagani et al75 ). Increased expression of SMO, as well as the downstream targets GLI1 and PTCH1, was noted in the human CML stem cell compartment as well as in BCR-ABL1+ cells in both chronic phase and blast crisis.76 Subsequent studies in the retroviral mouse model showed that Smo deficiency attenuated BCR-ABL1–induced CML-like leukemia and decreased the efficiency of secondary transplantation of the disease. Treatment with the SMO inhibitor cyclopamine caused significant prolongation of survival, a reduction in CML stem cells, and a reduction of disease onset in secondary transplantation recipients. Complementary to these results, the small-molecule SMO antagonist LDE225 (Novartis) caused a significant reduction in secondary colony formation and replating efficacy in primary CML cells in vitro, as well as improved survival in mouse models of CML.77,78 LDE-225 is in a phase 1b trial in combination with nilotinib for relapsed/refractory CML (www.clinicaltrials.gov identifier NCT01456676). Preliminary clinical results with another SMO antagonist, PF-04449913 (Pfizer; www.clinicaltrials.gov identifier NCT00953758) suggest significant activity against advanced CML and several other myeloid neoplasms.79 A third phase 1/2 trial of an oral SMO antagonist (BMS-833923) in combination with dasatinib for CML with suboptimal response to prior TKI therapy is currently on hold (www.clinicaltrials.gov identifier NCT01218477).
Targeting leukotrienes and prostaglandins
Arachidonate 5′-lipoxygenase (5-LO), the product of the Alox5 gene, catalyzes oxidation of arachidonate at the 5-position to yield 5-hydroperoxyeicosatetraenoic acid (5′-HPETE). 5-LO then converts 5-HPETE to leukotriene A4, leading ultimately to leukotriene B4, which mediates several physiological and pathological functions including inflammation, cancer, and neurodegeneration. Alox5 is up-regulated in CD34+ human CML cells,65 and previous studies showed that 5-LO inhibitors suppress proliferation and induce apoptosis of human CML cell lines.80 A recent study in the mouse retroviral CML model showed that in the absence of the Alox5 gene, BCR-ABL1 failed to induce CML-like leukemia. A small-molecule 5-LO inhibitor, zileuton, was more effective than imatinib in prolonging survival of mice with BCR-ABL1–induced CML-like disease and in combination with imatinib had a better therapeutic effect than either drug alone.81 Based on these findings, which suggest a role for 5-LO in CML stem cell survival and self-renewal, a clinical trial of zileuton (an FDA-approved asthma drug) in combination with imatinib was carried out in CML patients (www.clinicaltrials.gov identifier NCT01130688).
Deleterious effects on LSCs by another cyclooxygenase-derived metabolite, the cyclopentenone prostaglandin Δ12-PGJ3, were first predicted from an in silico screen for compounds that induce gene expression changes similar to the AML stem cell toxin parthenolide82 and were subsequently confirmed directly in the mouse retroviral model of CML.83 Nanomolar concentrations of Δ12-PGJ3 were selectively toxic in vitro to BCR-ABL1+ LSK cells, but not normal HSCs, whereas 1 week of daily intraperitoneal injections of Δ12-PGJ3 eradicated fatal CML-like leukemia in recipient mice and abolished secondary transplantation of the disease. Although the gene expression screen was based on a signature of NF-κB inhibition and induction of oxidative stress, the mechanism of BCR-ABL1+ stem cell killing by Δ12-PGJ3 did not appear to involve induction of reactive oxygen species, but rather activation of ATM and p53.83 Moving this exciting treatment strategy into the clinical setting might be complicated; although Δ12-PGJ3 can be produced endogenously from the dietary fish-oil component eicosapentaneoic acid, it is not clear that adequate tissue concentrations can be achieved through this route, possibly necessitating parenteral injections of the purified prostaglandin.
Modulating BCR-ABL1 stability
The chaperone HSP90 plays a role in regulating survival, proliferation, and apoptosis of cancer cells by acting as a chaperone for several client oncoproteins such as BCR-ABL184 and HER2. 17-Allylamino-17-demethoxygeldanamycin (17-AAG; tanespimycin) inhibits the binding of HSP90 to BCR-ABL1, resulting in down-regulation of BCR-ABL1 and apoptosis of CML cell lines.85 Interestingly, imatinib-resistant mutants of BCR-ABL1 are more sensitive to inhibition of HSP90 by 17-AAG than native BCR-ABL1.86 Two phase 1 trials of tanespimycin in refractory CML have been completed (www.clinicaltrials.gov identifiers NCT00066326 and NCT00100997), but further clinical development of the drug has been hindered due to poor solubility. IPI-504 (retaspimycin), a 17-AAG analog with better solubility, induces dissociation of BCR-ABL1 and HSP90 in BCR-ABL1–expressing cell lines as quickly as 30 minutes after treatment. In mice with CML-like leukemia induced by native or T315I mutant BCR-ABL1, IPI-504 treatment significantly prolonged survival and decreased the phenotypic LSC compartment.87 Clinical trials of retaspimycin in CML have not yet been opened.
Promyelocytic leukemia protein (PML) is a nucleolar protein best known for its role as a fusion partner for RARα in acute promyelocytic leukemia, but genetic studies in mice have revealed multiple roles for PML in regulation of apoptosis, cellular proliferation, and tumor suppression.88 A recent study demonstrated that PML is expressed in HSCs and at particularly high levels in CML stem/progenitor cells. In the mouse retroviral CML model, BCR-ABL1 expression in stem cells lacking PML induced leukemia in primary recipients but failed to propagate the disease in serial transplantations, indicative of a defect in LSCs.89 Treatment with arsenic trioxide, which targets PML for degradation and is in clinical use for therapy of acute promyelocytic leukemia, caused cell cycle entry of BCR-ABL1+ LSCs without inducing apoptosis, resulting in depletion of LSCs as assessed in LTCIC assays. In combination with cytarabine chemotherapy, arsenic treatment resulted in long-term survival of secondary recipients of BCR-ABL1–expressing stem cells.89 Subsequent studies suggested that the primary effect of arsenic on CML cells is destabilization of BCR-ABL1 through autophagosomal or proteasomal degradation.90,91 Although this is a promising therapeutic approach, there is some concern about toxicity because PML deficiency causes a defect in long-term repopulation by normal HSCs.89 Two clinical trials of arsenic trioxide in combination with ABL1 TKIs in CML patients with suboptimal response are underway (www.clinicaltrials.gov identifiers NCT00250042 and NCT01397734).
Inhibiting autophagy
Autophagy is a cellular reaction to metabolic stress characterized by increased breakdown of cell components and is mediated by intracellular formation of autophagosomes, which aids in cell survival. Some cancer cells use autophagy as a mechanism to avoid apoptosis or necrosis induced by chemotherapy or targeted inhibitors.92 When BCR-ABL1 kinase activity is inhibited by TKIs, CML cells induce an autophagy response through reactivation of an mTOR-ATF5 pathway that contributes to imatinib resistance. Inhibition of autophagy via knock-down of several essential autophagy genes or by compounds such as the antimalarial drug chloroquine increases the sensitivity of CML stem cells to killing in combination with TKIs.93 Chloroquine is currently in several clinical trials as an autophagy inhibitor in solid tumors and is being tested in combination with imatinib in a randomized phase 2 trial in CML patients with residual disease (www.clinicaltrials.gov identifier NCT01227135). Interestingly, another autophagy inhibitor, the antibiotic clarithromycin, has been shown to inhibit late-stage autophagy in CML cells94 and to accentuate the clinical response to TKIs in several patients with resistant CML.95
Targeting epigenetic regulators
Histone deacetylase inhibitors (HDACIs) are approved for treatment of cutaneous T-cell lymphoma and have been explored for activity in CML in several settings. The pan-HDACIs suberoylanilide hydroxamic acid96 and LAQ82497 down-regulated BCR-ABL1 levels and induced apoptosis in CML-mBC cells, whereas HDACIs potentiated the activity of ABL1 TKIs98 and inhibitors of polo-like kinase 199 against resistant CML cells in vitro and in vivo. More relevant to targeting CML stem cells, a recent study showed that LBH589 (panobinostat) induced apoptosis in quiescent CML progenitors that are resistant to elimination by imatinib and reduced NSG engraftment by these cells. In a conditional transgenic mouse model of CML, panobinostat in combination with imatinib also reduced BCR-ABL1+ LSCs.100 Based on these data, a phase 2 trial of panobinostat in refractory CML was completed (www.clinicaltrials.gov identifier NCT00451035), whereas a second phase 1 clinical trial of panobinostat in combination with imatinib in CML patients in cytogenetic remission with residual disease is under way (www.clinicaltrials.gov identifier NCT00686218).101
SIRT1 is an NAD(+)-dependent deacetylase that is overexpressed in human CML LSCs and activated by BCR-ABL1, promoting leukemogenesis.102 Pharmacologic inhibition (with resveratrol) or knock-down of SIRT1 increased apoptosis in LSCs from chronic phase and blast crisis CML and reduced their viability both in vitro and in a transgenic mouse model. SIRT1 effects were enhanced in combination with imatinib and were dependent on p53 expression and acetylation.103 Therefore, inhibiting SIRT1 with resveratrol or other small-molecule antagonists (niacinamide and SRT501) represents another potential strategy for targeting stem cells in CML, but this approach has not yet entered the clinical setting.
Targeting the BCL2 family
Hematopoietic stem cells express both pro- and antiapoptotic BCL2 family members, each with alternative splice isoforms. Whole-transcriptome RNA sequencing revealed that alternative splicing of multiple prosurvival BCL2 family members promotes the malignant transformation of myeloid progenitors in CML-mBC into LSCs that are largely quiescent in the BM niche and resistant to TKI therapy. The novel pan-BCL2 inhibitor sabutoclax (ONT-701) increased the sensitivity of mBC LSCs to TKIs at doses that spared normal progenitors.104 These findings suggest that the elimination of dormant LSCs could be achieved by combining TKI with inhibitors of the antiapoptotic BCL2 family proteins.
Another strategy for targeting short-lived BCL2 family members in CML is the use of agents that inhibit protein translation elongation. Omacetaxine mepesuccinate is a semisynthetic derivative of a plant alkaloid from Cephalotaxus that was recently approved for use in relapsed/refractory CML, including CML caused by the BCR-ABL1 T315I mutant. The predominant cellular mechanism of action of omacetaxine is inhibition of protein synthesis elongation through direct binding to the ribosome aminoacyl-tRNA acceptor site. In leukemia cell lines, omacetaxine treatment results in rapid declines in the levels of several short-lived proteins involved in apoptosis and cell proliferation, including MCL-1, Cyclin D1, and c-MYC.105 Exposure to omacetaxine at concentrations of 10 to 100 nM rapidly reduces MCL-1 levels and induces apoptosis of primary human CD34+ cells, but there is little difference in the sensitivity of normal and CML progenitors to the drug.106 For native (unmutated) BCR-ABL1, synergy between TKIs and omacetaxine has been demonstrated in CML cell lines107 and is suggested from the results of several pilot clinical studies.108,109 In the retroviral mouse model of CML induced by nonmutated BCR-ABL1, omacetaxine has been shown to prolong survival and decrease the abundance of phenotypic (GFP+LSK) CML stem cells.110 Due to its distinct mechanism of action that is independent of BCR-ABL1 kinase activity, omacetaxine is an attractive candidate that might be added to TKIs to deepen molecular responses in CML.
Immunological approaches to CML stem cell eradication
In the pre-imatinib era, allogeneic stem cell transplantation was the therapy of choice for CML and remains the only proven curative treatment. CML is one of the most immune-responsive neoplasms known and the majority of the benefit of allografting is thought to be due to a GVL effect mediated by allogeneic immune cells. The potency of GVL in CML was illustrated directly by the demonstration that transfusion of leukocytes from the allogeneic donor (donor leukocyte infusion) could induce remissions in the majority of CML patients who relapsed after allografting. Although the risks of transplantation have relegated this procedure to salvage therapy in the TKI era, the possibility of harnessing the immune system to eradicate CML stem cells is still an active area of investigation.
IFN-α
Before the advent of imatinib, IFN-α–based regimens were the pharmacologic treatment of choice in early phase CML, and several strategies to incorporate IFN-α into TKI therapy for CML have been tested. In the frontline setting, addition of pegylated IFN-α (peg-IFN-α) at a dose of 50 to 90 μg weekly to imatinib resulted in statistically significant improvements in MMR and CMR rates in independent randomized trials from France111 and Sweden.112 Although peg-IFN-α was discontinued in 45% to 60% of patients in the combination arm due to side effects, subgroup analyses suggest that even several months of IFN-α therapy significantly increased the molecular response rate to imatinib. A second approach is to add or continue IFN-α in TKI-treated patients with molecular residual disease. Although some have argued that the therapeutic effects of IFN-α are immune mediated and therefore patients derive maximum benefit from the drug when their leukemic burden is high (ie, at diagnosis), more recent evidence suggests that IFN-α can stimulate normal HSCs to proliferate,113 and if this extends to CML stem cells, might render them more sensitive to TKIs. A German study assigned 20 chronic phase CML patients to initial combination therapy with imatinib 400 mg/d and either recombinant IFN-α (n = 3) or peg-IFN-2-α (n = 17). After 2 years of combination therapy, imatinib was discontinued and molecular responses monitored while patients continued on IFN-α maintenance therapy. After a median of 2.4 years off imatinib, 10 of 15 evaluable patients had further decreases in BCR-ABL1 transcripts with the number achieving CMR increasing from 2 to 5 patients.114 Several clinical studies (www.clinicaltrials.gov identifier NCT00573378, University of Michigan; www.clinicaltrials.gov identifier NCT01392170, M.D. Anderson Cancer Center; and www.clinicaltrials.gov identifier NCT00297570, Sheba Medical Center) are currently testing the hypothesis that addition of IFN-α can enhance molecular responses in TKI-treated CML patients and perhaps increase the rates of CMR and possible cure.115
Vaccination strategies
Several studies over the past decade have explored whether vaccination of CML patients with various antigens can elicit immune responses against leukemic cells and correlate with improved clinical responses. Peptides derived from the BCR-ABL1 junctional region can elicit a peptide-specific T-cell immune response in CML patients.116 Larger observational studies of junctional peptide vaccination of chronic phase CML patients on imatinib (www.clinicaltrials.gov identifier NCT00466726) have demonstrated T-cell responses and reductions in BCR-ABL1 transcripts in vaccine recipients,117-119 whereas sustained CMRs in the absence of TKI therapy have been reported in a few vaccinated patients.120,121
Other antigens are also being tested in vaccine trials in CML. WT1 is an oncogene expressed by the Wilms tumor gene that is overexpressed in the majority of patients with CML but virtually absent in normal progenitors. WT1 vaccines prompt specific immune responses in patients with hematologic malignancies without significant side effects. When combined with imatinib, a WT1 peptide vaccine induced WT1-specific immune responses and was associated with achievement of CMR in a CML patient.122 PR1 is an immunogenic peptide derived from proteinase-3, a primary myeloid granule protein that is also expressed in stem/progenitor cells from patients with AML and CML.123 PR1-specific T lymphocytes can kill CML progenitors and their presence correlates with CCyR in CML patients treated with IFN-α.124 In a clinical trial of a combined PR1/WT1 peptide vaccine in myeloid neoplasms (www.clinicaltrials.gov identifier NCT00499772), low- and high-avidity CD8+ T cells were elicited, but repeated vaccinations were correlated with loss of high-avidity T-cell clones, suggesting a need for alternative vaccination schedules or approaches.125 Another strategy is the use of cell-based vaccines, such GVAX, which is based on the CML cell line K562 engineered to coexpress GM-CSF. In a clinical trial, GVAX vaccination was shown to reduce BCR-ABL1 transcripts in 13/18 recipients, 7 of whom achieved CMR.126 These studies demonstrate that vaccines are generally safe and that CML patients can generate T-cell responses while on TKI therapy and there are promising clinical responses in some vaccinated patients. However, randomized trials will be necessary to determine whether vaccination has benefits that are distinct from that of concurrent TKI therapy.
Novel cell surface target for immunotherapy
Although antibody-mediated immunotherapy (eg, with rituximab) has been very successful in B-cell lymphoma, its application to CML requires that CML stem cells express a specific cell surface antigen that is not expressed on normal HSCs. Although previous studies found the immunophenotype of CML stem cells to be identical to that of normal HSCs,18 a recent exciting study used a genomic approach to identify IL-1 receptor accessory protein (IL1RAP, also known as IL-1 R3) as an antigen specifically expressed on human Ph+CD34+CD38− cells that have some characteristics of LSCs, including LTCIC activity.127 The investigators further demonstrated specific natural killer cell-mediated killing in vitro of these Ph+ targets with a polyclonal anti-IL1RAP antibody and of AML stem cells by a monoclonal anti-IL1RAP antibody.128 However, because parallel xenograft experiments have not been done, there is no direct proof that IL1RAP specifically marks CML or AML stem cells or that IL1RAP can be exploited as an immunotherapy target in vivo.
Targeting the CML stem cell niche
A new frontier in targeting CML stem cells has emerged with the recognition that these cells exist in a BM niche that is distinct in several functional aspects from the normal HSC niche.129 As an attempt to overcome the quiescence of CML stem cells in the niche, treatment with myeloid cytokines increased the proliferation of quiescent CD34+ CML cells in vitro and enhanced the killing of these cells by imatinib.130,131 A key feature of such a clinical strategy might be the relative timing of cytokine and imatinib administration because acute exposure to imatinib tends to increase the quiescent LSC fraction even when cytokines are present. Based on this consideration, a clinical trial of continuous versus pulsed imatinib with or without G-CSF was launched.132 Stromal-derived cytokines, including IL-6 and G/GM-CSF133 and placental growth factor (PlGF)134 have also been shown to enhance BCR-ABL1–induced MPN and impair the therapeutic response to TKIs, providing motivation for the use of inhibitors of cytokine receptor or PlGF/VEGR1 signaling in CML. However, the response of BCR-ABL1–induced leukemia to dual treatment with imatinib and the JAK2 inhibitor TG101209 in the mouse retroviral CML model was disappointing, perhaps due to suppressive effects on normal hematopoiesis.135 Adhesive interactions of CML stem cells with niche stroma, including N-cadherin–mediated activation of β-catenin136 and activation of CXCR4 signaling by stromal expression of the chemokine CXCL12 (SDF-1),137 induce quiescence and TKI resistance that might be overcome by antagonists of these pathways such as the CXCR4 inhibitor plerixafor. Adding plerixafor to dasatinib did not reduce the leukemia burden in mice with BCR-ABL1–induced CML-like leukemia and was associated with infiltration of the CNS by leukemic cells, raising caution in testing this approach in CML patients.138 Finally, activation of the parathyroid hormone receptor in BM osteoblasts attenuates BCR-ABL1–induced CML-like leukemia, but enhances MLL-AF9–induced AML in mouse retroviral models, possibly through opposing effects of increased TGF-β1 on the respective LSCs. Parathyroid hormone (PTH) treatment of wild-type mice with CML-like leukemia caused a 15-fold decrease in LSCs and reduced NSG engraftment by primary human CML cells.139 These results suggest that pharmacologic manipulation of the CML stem cell niche is a novel strategy for eradicating these cells.
Conclusions and future directions: getting into the clinic
From the foregoing summary, it is clear that we have no shortage of innovative approaches to eliminating CML stem cells. A major challenge for the field is testing which of these strategies will be safe and effective in our CML patients who are on TKI treatment, recognizing that most of these patients are asymptomatic with an excellent quality of life and, as a corollary, that any proposed intervention must have very low toxicity and morbidity. The time is ripe for clinical trials of these approaches because we now have multiple targeted therapies with dosing and safety profiles as single agents that have been established. Our own preferences for those strategies that have the strongest existing evidence and best chance for success are indicated in Table 1. Because the depth of molecular responses continues to increase with time on TKI treatment, it is clear that such trials will need to be randomized and must compare cohorts of patients on the same TKI at the same point after initiation of treatment. Given that CML is a relatively rare condition, this poses a challenge, but the rising prevalence of the disease and the potential cost savings of discontinuing TKI treatment should motivate all stakeholders (patients, caregivers, payers) to participate in such studies. Outside of a clinical trial, should we advise our CML patients to drink wine, consume fish oil, or take NSAIDs? That these are all reasonable suggestions is a measure of just how far we have come in the treatment of CML.
Acknowledgments
This work was supported in part by the National Institutes of Health (Grants T32 CA009429 to W.A. and R01 CA090576 to R.A.V.). The authors thank Drs Daniela Krause and John Goldman for helpful comments on the manuscript and apologize to those researchers whose work was not cited due to length considerations.
Disclosures
Conflict-of-interest disclosure: R.A.V. has received research funding from TEVA Pharmaceuticals and has consulted for Bristol Myers Squibb, Deciphera Pharmaceuticals, and TEVA Pharmaceuticals. W.A. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Richard A. Van Etten, MD, PhD, Chao Family Comprehensive Cancer Center, University of California, Irvine, 839 Medical Sciences Court, Sprague Hall, Room 124, Irvine, CA 92697-4047; Phone: 949-824-2655; Fax: 949-824-4023; e-mail: vanetten@uci.edu.
