
Abstract
Natural killer (NK) cells represent a key component of innate immunity. The utility of mouse models to recapitulate the human immune response has been a matter of ongoing debate, especially with regard to NK cells. However, mouse models of NK cells have provided significant advancements in our understanding of the biology of the cells that bridge these species. Initial characterization of NK cell activity was in mouse hematopoietic stem cell transplantation models. Recent findings include uncovering functionally disparate subsets of NK cells based on unique inhibitory receptor expression patterns, the existence of memory-like NK cells, and immunoregulatory NK cells that affect hematopoiesis and T-cell function. In addition, the biology of these cells with regard to MHC-binding receptors that affect NK cell subset maturation and function in the context of licensing, the importance of cytokines such as IL-15 in their development and maintenance, and evidence of NK exhaustion have been initially studied in mice. Many of these findings have been validated in clinical studies and demonstrate the significant wealth of knowledge that can be obtained by mouse models. However, it is important to understand the limitations and conditions of the mouse models, particularly when studying NK cells in hematopoietic stem cell transplantation and cancer.
Introduction
Natural killer (NK) cells represent a critical component of innate immunity aiding in the defense against malignant and virally infected cells. There has been recent concern about using mice to model inflammatory human conditions due to transcriptional response differences between the two species.1 However, looking back on the progression of science and the study of immunology, mouse models have dramatically shaped our understanding of the fundamental basis of immune responses to various disease states and helped to delineate the complexities of the immune system in an in vivo setting. Some areas of study in immunology that have greatly benefited from the use of mouse models are hematopoietic stem cell transplantation (HSCT) and NK cell biology. Taking into account the differences and limitations of mouse models, a more thorough understanding of the similarities between humans and mice has resulted in significant progress on the fundamental nature of NK cell function, differentiation, and regulation, particularly in the context of HSCT.
NK cell activity was first reported in 1964 through the observation of spontaneous bone marrow (BM) allograft rejection by lethally irradiated unsensitized host mice. This was followed by the discovery of hybrid resistance, the capability of irradiated F1 hybrid mice to reject the parental BM allografts but not solid tissue allografts, which was at variance with the laws of transplantation. Finally, the observation of MHC-unrestricted killing of tumor targets by human NK cells in vitro was reported and the field of NK biology was born. Subsequent in vivo studies using mouse models demonstrated that these cells were prominent in viral resistance and resistance to malignant cell types, particularly those of hematopoietic origin or solid tumors undergoing metastasis.
NK cells differentiate from the common lymphoid progenitor in the BM and enter circulation after maturation, with minor differences in development between NK cells from humans and mice (Figure 1). As part of the innate immune system, they provide rapid responses to viral infections and tumors by lysing targets via release of perforin and granzyme and by producing various cytokines including IFNγ, TGFβ, TNFα, IL-6, IL-10, GM-CSF, and G-CSF. Studies using viral models, HSCT, and tumor models in mice have uncovered nuances of NK cell biology beyond the initial simplistic cell types they appeared to be. The models use a variety of techniques to uncover NK function and biology, including adoptive transfer of NK cells, lethal irradiation, and allogeneic BM cell transfer, monoclonal antibody depletion of particular subsets of NK cells or other cells, antibodies against tumor targets to study antibody-dependent cell-mediated cytotoxicity, and cytokine administration to stimulate and enhance NK activity. These models have uncovered characteristics of NK cells that include a diverse array of NK subsets with differential functions, positive selection during development, memory-like NK cells that may be long lived, and potent immunoregulatory roles of these cells that may actually be their predominant role. Several of these characteristics have been validated and applied in clinical studies and will be described throughout the course of this review, along with the trepidations and issues concerning mouse models.

NK cell development and maturation in humans and mice. Early stages of natural killer (NK) cell development are identical between humans and mice with the hematopoietic stem cell that progresses to the common lymphoid progenitor (CLP) with c-KIT, FLT3, and IL-7R expression. The CLP then differentiates into the NK progenitor (NKP) that is marked by expression of IL-2Rβ (CD122). As the NK cell continues to develop, differences arise in the markers between humans and mice, but analogous receptors are expressed at each stage by the NK cells. In the immature NK cell stage that develops from the NKP, CD2 is expressed in both humans and mice with the addition of the NK-cell receptor protein 1 molecule (NK1.1 in many strains of mice or CD161 in humans). The next stage is the immature lytic NK cell, where human NK cells start to express CD56 and mouse NK cells express DX5. This is followed by expression of CD94 in both humans and mice. The final, mature lytic stage of NK cells is determined by the expression of receptors that are capable of binding to MHC-I and related molecules: the Ly49s in mice and the killer cell immunoglobulin-like receptors in humans. The NK cells can now leave the bone marrow and head out into the periphery in their mature state.40
NK cell development and maturation in humans and mice. Early stages of natural killer (NK) cell development are identical between humans and mice with the hematopoietic stem cell that progresses to the common lymphoid progenitor (CLP) with c-KIT, FLT3, and IL-7R expression. The CLP then differentiates into the NK progenitor (NKP) that is marked by expression of IL-2Rβ (CD122). As the NK cell continues to develop, differences arise in the markers between humans and mice, but analogous receptors are expressed at each stage by the NK cells. In the immature NK cell stage that develops from the NKP, CD2 is expressed in both humans and mice with the addition of the NK-cell receptor protein 1 molecule (NK1.1 in many strains of mice or CD161 in humans). The next stage is the immature lytic NK cell, where human NK cells start to express CD56 and mouse NK cells express DX5. This is followed by expression of CD94 in both humans and mice. The final, mature lytic stage of NK cells is determined by the expression of receptors that are capable of binding to MHC-I and related molecules: the Ly49s in mice and the killer cell immunoglobulin-like receptors in humans. The NK cells can now leave the bone marrow and head out into the periphery in their mature state.40
Differences and similarities between human and mouse NK cells
Although significant differences exist between NK cells found in humans and mice (Table 1) that could be attributed to species divergence or the fact that research mice are inbred and maintained under specific-pathogen-free conditions, many of the fundamental principles of NK biology and function can be studied in mice and applied to humans. One key area of NK divergence between human and mouse NK cells lies with the receptors they express. The inhibitory receptor NKG2A and the activating receptor NKG2D that binds stress ligands (in humans: MICA, MICB, ULBPs; in mice: Rae-1, H-60, MULT1) are similar in terms of structure, function, and expression between humans and mice. However, the killer cell immunoglobulin-like receptors (KIRs) in humans and the C-type lectin-like family of receptors, Ly49s, in mice differ greatly in their structures and binding, but are similar in their signaling pathways, functions in terms of inhibition and activation (some receptors are inhibitory when bound to MHC class I (MHC-I), others are activating when bound to their ligands), and involvement in NK functionality that will be discussed below.
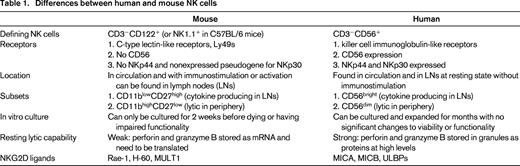
In contrast to the similarities mentioned above, the marker CD56 is specific to human NK cells and is not present on mouse NK cells. CD56 is of great significance because of its differential expression denoting 2 distinct subsets of NK cells in terms of function and location within the body. The CD56dim population consists of highly cytotoxic cells with low levels of cytokine production and is primarily located in the periphery. The CD56bright population, contrastingly, is poorly lytic, produces high levels of cytokines, and is found primarily in the lymph nodes.2 The presence of NK cells in the lymph nodes is another significant difference between human and mouse NK cells, because human NK cells can be found normally in the lymph nodes, whereas NK cells are not found there in nontreated or nonmanipulated mice and are found there only after stimulation.3 Correlations between CD11blowCD27high and CD11bhighCD27low NK cells in mice with CD56bright and CD56dim, respectively, can be made in terms of functionality and migration.4 These differences would indicate that, although the function of this NK cell subpopulation in the lymph nodes is as yet unclear, NK cells may be evolving for more immunoregulatory functions in addition to directly lytic innate effects.
Further differences can be seen in cultured human versus mouse NK cells that exhibit dramatically different initial functionality and cytotoxicity. Human NK cells exhibit strong cytotoxic capabilities when newly isolated from the peripheral blood and can be maintained and expanded in culture for months. These cells can be grown with IL-2 and/or IL-15 for the initial few weeks, but long-term culture requires feeder cell populations to help maintain NK cell proliferation and activity for months.5 Murine NK cells exhibit weak lytic capabilities when freshly isolated and can only be maintained in culture with IL-2 for 2 weeks, after which time cell death and measurable decreases in cytotoxic function and cytokine production occur.6 The differences in initial cytotoxic capabilities can be attributed to the differences in perforin and granzyme B translation and protein expression. In mice, perforin and granzyme B mRNA are constitutively transcribed, but minimal levels of protein are detected until stimulation or activation of the NK cells that leads to rapid translation.7 However, human NK cells have high levels of perforin and granzyme B protein constitutively present that results in their high basal level of cytotoxicity and rapid response.8 It is unclear whether these differences can also be attributed to specific-pathogen-free housing conditions of inbred mice.
Although the differences in human and murine NK cells may appear numerous and may seem to outweigh the usefulness of the mouse model (and potentially highlight an evolutionarily later development than the more conserved, yet seemingly more advanced T cells), many of the findings regarding NK cells, including ontogeny, function, and regulation, that were initially observed in mouse studies have been validated with human NK studies. Furthermore, there are distinct advantages of using the mouse as a model. Mice allow for the modeling of complex disease states in vivo. The vast array of reagents, technologies, and genetic knowledge developed for mice allow for the study of processes and dissection of mechanisms and therapies using NK cells that cannot be performed using larger outbred animals because of the lack of transplantable tumors for these animals. Therefore, the limitations and issues concerning mouse models and the differences that exist between human and mouse NK cells should be acknowledged and noted, but by designing thoughtful and representative experiments that can address the underlying mechanisms, functions, and principles of NK cell biology, many of the differences can be accounted for and applied to humans (Table 2). The following sections will highlight studies and findings done in mouse NK cells that can be applied to and validated in humans.
NK cell education and licensing
NK cell development and ontogeny is one of the most significant aspects of NK biology to which mouse studies have contributed. It is now thought that NK cells go through a process analogous to positive selection of thymocytes in the thymus whereby NK cells need to be able to detect and bind to self to become fully functional. The KIRs (in humans) and Ly49s (in mice) include inhibitory receptors with different binding affinities to MHC-I haplotypes.9 It has been proposed that only the NK cells that express specific inhibitory receptors with high binding affinities to the MHC-I haplotypes present are fully functional and possess lytic and cytokine producing capabilities. These cells are termed “educated” or “licensed.” The cells that do not possess inhibitory receptors capable of binding to self are termed “unlicensed” or “uneducated” and show decreased production of IFNγ and reduced cytotoxic capabilities in vitro (Figure 2).10,11

Process of NK cell licensing. Shown is the process of NK cell licensing based on research by Kim et al.10 If a NK cell possesses an inhibitory receptor capable of binding to self-MHC-I, it becomes a licensed, fully functional NK cell that can be inhibited. However, if it does not possess an inhibitory receptor capable of binding to self, it is unlicensed, hyporesponsive, and thus does not need inhibition.
Process of NK cell licensing. Shown is the process of NK cell licensing based on research by Kim et al.10 If a NK cell possesses an inhibitory receptor capable of binding to self-MHC-I, it becomes a licensed, fully functional NK cell that can be inhibited. However, if it does not possess an inhibitory receptor capable of binding to self, it is unlicensed, hyporesponsive, and thus does not need inhibition.
Initial in vivo studies looking at functional differences between licensed and unlicensed subsets in mice were conflicting. Some studies supported the in vitro models with the licensed NK cells rejecting allogeneic BM cells (BMCs), including those from MHC-I knockout mice, indicating that the differences in rejection observed were not due to potential inhibition by MHC-I recognition.12 This was one of the first studies showing in vivo functional differences between licensed and unlicensed NK cells, and it has significant clinical ramifications with the identification of the particular subset of cells that mediate BMC rejection after HSCT. However, a different study looking at differences between the subsets in murine CMV (MCMV) resistance showed that depletion of the unlicensed population resulted in significantly greater viral titers after infection compared with depletion of the licensed population. The researchers concluded that any benefit in functionality obtained by licensing was outweighed by the inhibition that occurred because the licensed cells could actually recognize self and be inhibited.13 A recent study demonstrated that the licensed population is greatly regulated and suppressed by T-regulatory cells (Tregs) because, after HSCT or after Treg depletion, depletion of the licensed population resulted in far greater viral loads than depletion of the unlicensed population, especially early during the course of MCMV infection.14 This suggested that licensed cells are highly regulated to prevent overactivation and damage to self. Based on these studies, licensed NK cells appear to play a large role after HSCT (when NK cells are more radioresistant than other lymphocytes and repopulate faster as well) and mediate BMC rejection and viral protection. In addition, recent studies demonstrated regulation of NK cells by Tregs through control of IL-2 availability that affected NK cell activity and homeostasis in mice,15 further demonstrating the capability of Tregs to regulate NK cells. Myeloid-derived suppressor cells have also been shown to regulate and inhibit NK cell activity in patients with hepatocellular carcinoma,16 suggesting the existence of the multifaceted regulation that exists for NK cells.
Clinical studies corroborated these findings on licensing in patients who had undergone a HSCT and had CMV reactivation. These patients showed an increased and sustained expansion of licensed, NKG2C+ NK cells that also produced increased levels of IFNγ, indicating that the licensed subset of NK cells may have a more significant role after HSCT, when patients are immunocompromised with reduced Treg populations.17 Interestingly, a study looking at the shaping of the NK repertoire after various infections showed that infection by CMV promoted the expansion of licensed NK cells with self-specific KIRs, highlighting the preferential expansion of licensed cells in response to virus.18 Therefore, licensing of NK cell subsets based on inhibitory receptor expression appears to occur in vivo. However, a recent clinical study demonstrated that unlicensed NK cells exhibited greater antibody-dependent cell-mediated cytotoxicity in neuroblastoma patients given anti-disialoganglioside GD2 mAb. They concluded that, similar to the study by Orr et al on MCMV in mice,13 the licensed cells were inhibited by the high expression of MHC-I that was maintained in the tumor targets.19 The presence of Tregs and immunosuppression by the tumor microenvironment may also contribute to the observed results. Therefore, our understanding of the phenomenon of licensing and the disease states and immune conditions where it may be most significant is still limited. New studies are identifying new ligands for Ly49 receptors that appear to be involved in licensing (the nonclassical MHC-I molecule H2-M3 by Ly49A).20 In addition, NKG2A and other molecules such as 2B4 likely also contribute to licensing. The physiologic role of licensing likely relates more to the kinetics of responses and adds layers of protective pathways to control NK cell responses, because it is likely that both licensed and unlicensed subsets contribute to the net response by NK cells.
NK cell subsets
Beyond categorizing NK cells into subsets based on CD56 expression levels in humans or on licensing and the capability of self-recognition by NK cells, recent work in mice and humans has identified other markers for delineation of subsets with differing functional capabilities. Delahaye et al showed that the activating receptor NKp30, the ligands of which were thought to be the HLA-B-associated transcript-3 (BAT-3) and B7-H6, actually had 3 unique isoforms, with one of the isoforms having a separate signaling pathway.21 By looking at gastrointestinal stromal tumors, Delahaye et al found that NK cells that expressed 2 of the isoforms of NKp30 had the expected outcome of mediating NK cell activation and lysis of the tumor targets, whereas the third isoform resulted in NK cells producing immunosuppressive cytokines such as IL-10.21
Recent work on a different activating natural cytotoxicity receptor, NKp46, which is found on both mouse and human NK cells, suggests the potential of different NK subsets and various isoforms of NKp46 as well. Studies have suggested that influenza hemagglutinin is a ligand for NKp46.22 In contrast, another study looked at the role of this receptor through NKp46-knockout mice and showed improved viral control through T-cell regulation in the knockout mice compared with wild-type.23 Using these knockout mice and having the tools and reagents to try to determine other potential ligands and isoforms of the receptor, a better understanding of this receptor and the potential subsets of NK cells that express it can be ascertained.
NK cell immunoregulatory capabilities
An often-neglected aspect of NK biology and function is the role of NK cells with regard to immunoregulation and the link that NK cells provide between the innate and the adaptive immune system. Previous studies have demonstrated NK cells directly lysing infected dendritic cells (DCs), which in turn limits the number of antigen presenting cells (APCs) capable of activating and presenting to the adaptive immune cells.24 This reduces not only DCs, but also the antigen-specific T-cell populations. In addition, NK cells and DCs can activate one another, especially through the trans-presentation of IL-15 by DCs to activate NK cells.24,25 Recent studies have begun to demonstrate a more direct role of NK cells to regulate and suppress the adaptive immune response after viral infection. Waggoner et al showed that NK cells are capable of directly lysing CD4+ T cells, which in turn reduces the CD8+ T cells in lymphocytic choriomeningitis and influenza infections.26 They demonstrated that at low and medium doses of the virus, this reduction in T cells impaired the antiviral response and led to increased harm to host mice. However, at high doses of the virus, the regulation of T cells by NK cells was beneficial, with reduced immunopathology in target organs by the overactive T cells.26 Therefore, NK cells were shown to play a role in helping prevent immunopathology due to overactive T-cell responses after viral infection in mice.
More studies investigating the direct mechanism of this T-cell regulation by NK cells, the specific subsets of NK cells and cytokines involved, and characterizing the implications of this regulation in humans and various diseases need to be conducted because these findings were primarily demonstrated through mouse models. There have been studies using human cells that have shown similar lysis and regulation of T cells by NK cells in hepatitis B virus27 and human parainfluenza virus type 328 that supported these findings in mice, but the clinical implications of this NK-mediated inhibition of T cells has not been demonstrated in humans. However, the clinical implications of these findings are still numerous in both viral and cancer disease states in terms of both trying to amplify the adaptive immune response to aid in eliminating the disease state and also to shut off the response, especially in influenza cases, where the adaptive immune cells lead to severe immunopathology that can be lethal. There are numerous targeted clinical therapies that could use this immunoregulation by NK cells, including immunotherapies to enhance NK activity and function. Administration of IL-2, IL-15/IL-15Rα, or lenalidomide can be used to enhance NK regulation and potential lysis of T cells and help to reduce various autoimmune diseases and exaggerated immune responses such as those that occur during infection with highly pathogenic strains of influenza. Blocking death ligands or NKG2D ligands may prevent lysis of T cells by NK cells. In addition, decreasing NK function by targeting inhibitory KIRs can improve immune responses. However, suppressing the NK response may significantly dampen the overall immune response to various diseases and cancers, so more studies are needed to determine the optimal perturbations and kinetics to tune NK functionality to result in the ideal immune response.
Memory NK cells
A recent, novel finding in NK cells involves the existence of memory-like NK cells with expansion of particular subsets that persist at an elevated state for extended periods of time. Studies have shown NK cells that were induced by hapten-specific contact hypersensitivity and cytokine stimulation had an amplified response to secondary stimulation or when transferred to a new host mouse.29 Expanding on these findings, reports have shown that, after MCMV infection, there is an expansion of the Ly49H+ (an activating receptor that binds to the MCMV glycoprotein m157) NK cells that persists for months and leads to an amplified response to secondary infection or after transfer to a new mouse with a primary infection.30 These findings in mice of memory-like NK cells were soon studied in humans to determine whether a similar phenomenon occurred. Clinical studies identified that the NKG2C+ NK cells expanded and persisted for months after CMV infection, identical to what was discovered in mouse studies.17 These cells could potentially provide enhanced antiviral resistance, especially after HSCT, when CMV reactivation is a significant risk. These cells were in fact shown to be transplantable after HSCT from CMV-seropositive donors, greatly expanded and provided enhanced anti-CMV protection in recipients after viral reactivation, and showed enhanced cytokine production in response to target cells.31 Therefore, this memory NK population is of significant clinical importance to help reduce complications from CMV reactivation after HSCT. These findings can be further developed and these memory NK cells further defined, including other potential targets for specific memory cell populations beyond CMV, through a combination of mouse and human studies for potential clinical uses.
Clinical utilization of NK cells
The potential for therapeutic utilization of NK cells is promising. Two different approaches are being investigated, including cytokine administration to stimulate host NK cells and adoptive transfer of new NK cells. IL-2 therapies for stimulation have proven problematic, with severe toxicities including vascular leak syndrome, which leads to pulmonary edema and cardiovascular failure. IL-2 also leads to an expansion of Tregs that can negate the activation of the NK cells.32 Preclinical mouse studies showed the importance of the trans-presentation of IL-15 by DCs to NK cells to induce activation.25 Because of these mouse studies, IL-15/IL-15Rα complexes that mimic this trans-presentation have been tested in both humans33 and mice34 and were shown to activate and enhance the function of NK cells and to avoid the complications from IL-2 administration. Another study used K562 cells that had been transduced to express IL-15/IL-15Rα to expand and activate human NK cells ex vivo for future adoptive transfer of these cells.35
Adoptive transfers of NK cells have shown limited clinical benefit in terms of antitumor responses. Although NK cells have been shown to successfully engraft, expand, and migrate to tumor sites after adoptive transfer,36 clinical trials showed mixed results ranging from no changes in patient survival or metastasis occurrence to slight improvements.36,37 A study by Gill et al offered a possible explanation for the limited efficacy of the adoptive transfer of NK cells by demonstrating NK exhaustion as occurring after prolonged exposure to the tumor using mouse models.38 The investigators demonstrated that the transferred NK cells were capable of migrating to tumor targets and were highly lytic, with antitumor capabilities and cytokine production the first day after transfer. However, starting at day 5 after transfer, there was marked reduction in IFNγ production, cytotoxicity, and activating receptor expression, making the cells predominantly hyporesponsive.38 This suggested that the NK cells are capable of reaching their targets and mounting an antitumor response, but they become rapidly exhausted and hyporesponsive, thus limiting the clinical benefits. NK cell adoptive transfer may have the greatest benefits after allogeneic HSCT based on a prior study showing that adoptive transfer of activated NK cells of donor origin after allogeneic HSCT cannot only reduce GVHD, but also improve graft-versus-tumor effects in mouse models.39 These adoptive transfers have been performed in haploidentical settings where the partial HLA disparity can actually be beneficial in terms of NK enhanced reactivity due to the lack of inhibition by tumor targets. The utilization of specific subsets of NK cells, potentially the licensed population, with greater effector functions, may enhance these observed effects and improve clinical outcomes of patients undergoing allogeneic HSCT for cancer therapy.
Conclusions
Improving the clinical utilization of NK cells may dramatically improve antitumor therapies and reduce the life-threatening viral infections that commonly occur after immunosuppression from HSCT. Recent mouse studies have dramatically expanded our understanding of the biology and function of NK cells, including information on subsets of NK cells, how NK cells gain functionality, immunoregulatory roles of NK cells and memory NK cells, and NK cell exhaustion. These novel findings were aided by the flexibility and manipulation allowed in mouse models and studies (Table 3). There are considerable differences between mouse and human NK cells, including receptor expression and location throughout the body, but by acknowledging these differences and designing experiments that incorporate them, significant advances in NK biology can, and have, been made. Therefore, information on the sophistication of NK cells that has been uncovered so far through preclinical mouse studies can now be used and verified in clinical studies. By identifying specific subsets of NK cells that are truly the effector population that is desired (be it antitumor or immunoregulatory) and finding ways to limit NK exhaustion or use expanded memory NK cells, the therapeutic utilization of NK cells will improve dramatically. It is through such a combination of mouse and human studies that science and clinical practice will ultimately advance.

Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Dr William J. Murphy, Professor and Chair of Dermatology, Departments of Dermatology and Internal Medicine, UC Davis School of Medicine, University of California, Davis, 2921 Stockton Blvd, IRC Bldg, Rm 1614, Sacramento, CA 95817; Phone: 916-703-9397; Fax: 916-703-9396; e-mail: wmjmurphy@ucdavis.edu.
