
Abstract
For more than a century, the concept of a “magic bullet” to deliver cytotoxic therapy to the site of disease has been envisioned but only recently have technological advances enabled antibody-drug conjugates to fulfill that dream. The recent approvals of brentuximab vedotin and ado-trastuzumab emtansine and emerging data for many molecules in clinical trials highlight the potential for antibody-drug conjugates to offer new therapeutic options for patients. This chapter reviews the evolution, state of the art, and potential future improvements that are enabling rapid development of this important class of cancer therapeutics.
Introduction
The term “magic bullet,” attributed to the German physician Paul Ehrlich more than a century ago, captures the deceptively simple concept that a targeting agent could be harnessed to deliver cytotoxic therapy directly to the source of disease.1 This presaged the beginning of antibody-drug conjugates (ADCs) by many years. However, at the time, the tools that would be needed to make this dream a reality did not exist. It was not until the advent of monoclonal antibody technology in the mid-1970s that therapeutic antibodies against relevant cancer targets were developed and began to be tested.2 Since then, the era of molecular biology allowed such important advances as humanization and engineered cell lines to produce vast quantities of biological reagents. The discovery of potent cytotoxic agents and the chemistry that enabled conjugation to protein and linkers were applied to early antibodies. However, it would require decades of additional experimentation through trial and error before the right elements were combined to create the first approved ADC, gemtuzumab ozogamicin (GO), for the treatment of relapsed acute myeloid leukemia (AML).3
GO: is there a second act?
GO received accelerated approval based on 3 uncontrolled phase 2 trials demonstrating antitumor activity in patients with relapsed AML (13% complete responses and 13% complete remission with incomplete platelet recovery).4 However, a confirmatory trial in frontline patients failed to demonstrate a benefit and GO was withdrawn in June 2010.5 Since then, multiple additional randomized trials have demonstrated that patients with good-risk cytogenetics benefit when GO is combined with standard of care induction chemotherapy.6-9 There are several technical challenges associated with GO, including variable drug loading per antibody, unstable linker technology, and susceptibility to common resistance mechanisms in AML. Despite these shortcomings, learning how to best use GO as part of a frontline combination regimen has rekindled interest in making this early ADC available again for patients.10
ADCs come of age
The current technology has evolved significantly, resulting in the recent clinical success of 2 next-generation ADCs: brentuximab vedotin (ADCETRIS), targeting CD30 on relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma,11 and ado-trastuzumab emtansine (Kadcyla), targeting Her2+ breast cancer.12 In the case of ADCETRIS, accelerated approval in August 2011 was based on 2 single-arm open-label trials demonstrating unprecedented antitumor activity in patients with relapsed or refractory Hodgkin lymphoma (75% objective response rate, 34% complete response) and systemic anaplastic large cell lymphoma (86% objective response rate, 56% complete response).13,14 Trastuzumab emtansine received approval in February 2013 for patients with metastatic Her2+ breast cancer who failed prior therapy with trastuzumab and a taxane. Approval was based on a randomized trial of Kadcyla versus lapatinib plus capecitabine, demonstrating an increase in progression-free survival (hazard ratio = 0.65; P < .001; median progression-free survival 9.6 vs 6.4 months) and overall survival (hazard ratio = 0.68; P < .001; median survival 30.9 vs 25.1 months).15 With these 2 novel agents, the development of ADCs has entered a new phase, characterized by a large number of promising molecules in clinical testing and a significant investment in trying to refine the science and technology that underlies this powerful therapeutic modality.
ADCs: pharmacokinetic advantage versus chemotherapy
Traditional chemotherapy employs potent small molecules to kill rapidly dividing cells, often through antimitotic or DNA-damaging mechanisms. Systemic administration of these drugs results in both tumor killing and damage to healthy tissues. The balance between these 2 actions limits the efficacy and tolerability of single-agent chemotherapy. As a result, most cancer regimens consist of combinations of chemotherapy agents, each administered at or near the maximum tolerated dose and for a limited duration due to uptake and cumulative damage to normal tissues.16 The rapid clearance of small molecules and increased hydrostatic pressure in solid tumors further decrease the tumor-specific activity of chemotherapy.
In contrast, monoclonal antibodies, are large molecules (150 kDa) that are retained in the vasculature for several weeks and slowly diffuse into perivascular tissue.17,18 The complementarity-determining regions can provide high-affinity binding directed against cell-surface antigens on tumor cells. The combination of a long half-life, specificity for tumor cells, and high affinity results in the accumulation of antibody at the tumor over time. The lack of direct cytotoxicity often allows prolonged treatment that is well tolerated. However, most monoclonal antibodies have limited single-agent activity against cancer and are frequently used in combination with chemotherapy. Despite decades of active development, only 9 naked antibodies directed at 6 molecular targets are currently approved by the US Food and Drug Administration (FDA) for cancer therapy (Table 1).17
Unconjugated monoclonal antibodies approved for cancer
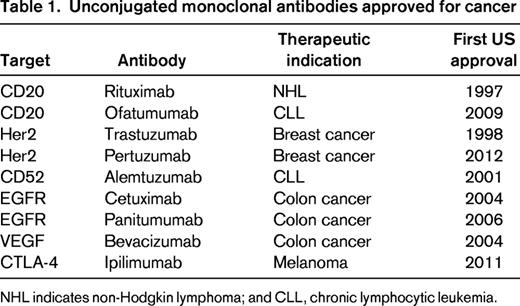
NHL indicates non-Hodgkin lymphoma; and CLL, chronic lymphocytic leukemia.
ADCs are designed to take advantage of the potent cell-killing activities of small molecules and the pharmacokinetic and biodistribution properties of monoclonal antibodies.19 ADCs empower antibodies by chemically conjugating a cytotoxic payload that can be delivered and released at the tumor while limiting systemic exposure to the cytotoxic agent. As shown in Figure 1, the mechanism of action for an ADC includes antibody engagement with a cell-surface target on cancer cells, internalization and intracellular trafficking of the intact macromolecule to the lysosome, rapid release of the cytotoxic agent, and efficient killing of tumor cells. In this chapter, several of the recent chemical and biological advances are highlighted that have taken ADCs from the laboratory to clinic and now into the forefront of the battle against cancer.

Mechanism of action of ADCs. The ADC binds to tumor cell surface antigens and is taken up within cells by a process known as receptor-mediated endocytosis. Upon entry into degradative compartments such as lysosomes, the drug can be released by linker hydrolysis or antibody degradation. Cell death occurs once the drug binds to its target.
Mechanism of action of ADCs. The ADC binds to tumor cell surface antigens and is taken up within cells by a process known as receptor-mediated endocytosis. Upon entry into degradative compartments such as lysosomes, the drug can be released by linker hydrolysis or antibody degradation. Cell death occurs once the drug binds to its target.
There are 5 important elements in the design of effective ADCs: (1) the molecular target; (2) the delivery vehicle (monoclonal antibody or alternative scaffold); (3) chemical conjugation (method, site, and stoichiometry); (4) the linker, including mechanism of drug release; and (5) the cytotoxic agent or payload.20,21 Current concepts for each of these elements are addressed in this review.
Choosing a good target
Several attributes have been proposed for an ideal ADC cancer target.20 These include abundant and uniform expression on the cell surface of tumor cells, low/absent expression on healthy tissues (especially vital organs), rapid internalization and trafficking to lysosomes upon antibody engagement, and accessibility to intravascular macromolecules. Unfortunately, there are a limited number of targets that fulfill all of these criteria, and clinical scientists must choose which qualities of the target antigen are most desirable. The 3 validated ADC targets (CD30, Her2, and CD33) each fulfill most of the proposed criteria. For CD30, high, uniform antigen expression on certain lymphomas, restricted normal tissue expression (limited to immune cell subsets), and rapid internalization made this an attractive ADC target. In the case of Her2, the target was already validated by the approved antibody, trastuzumab, and companion diagnostic tests were available to select patients with high antigen expression on tumor cells. Although CD33 is expressed at lower antigen density than either CD30 or Her2 on target tumor cells, it is uniformly expressed on almost all AML cells. Furthermore, the location of leukemia within the BM and intravascular space results in excellent antigen engagement for macromolecules.22 There are a large number of cancer targets that are currently being evaluated using clinical stage ADCs (Table 2). Over the next several years, results from these clinical trials will help to answer important questions about the choice of an ideal target for antibody-directed drug delivery. For example: How much antigen expression is required on tumor cells? How much expression on healthy tissue is tolerable? To what extent does antigen heterogeneity on tumor tissue and variable tissue penetration affect ADC antitumor activity? Some of these answers may vary based on tumor characteristics (solid tumor vs hematological malignancy, vascularity, and presence of a stromal barrier). The diversity of ADCs being tested currently will help to clarify many of these questions.
ADCs in clinical development
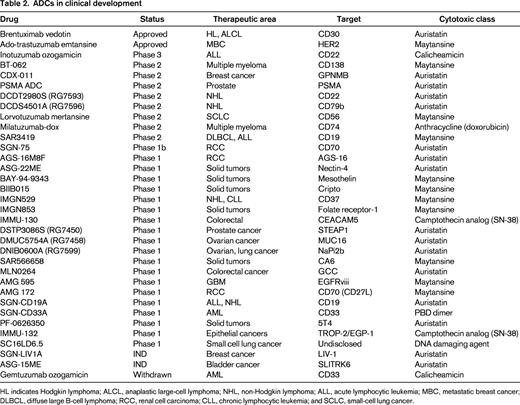
HL indicates Hodgkin lymphoma; ALCL, anaplastic large-cell lymphoma; NHL, non-Hodgkin lymphoma; ALL, acute lymphocytic leukemia; MBC, metastatic breast cancer; DLBCL, diffuse large B-cell lymphoma; RCC, renal cell carcinoma; CLL, chronic lymphocytic leukemia; and SCLC, small-cell lung cancer.
Selecting an optimal ADC antibody
The monoclonal antibody represents more than 90% of the mass of most ADCs in current development. The size and trafficking of an ADC play major roles in establishing the long half-life, which has been reported to range between 2 days and 2 weeks. Although the terminal half-life may be shorter than that of a naked antibody, a major impact of creating an ADC is to change the pharmacokinetics of the cytotoxic agent. The benefit of using fully human antibodies, as opposed to chimeric and humanized versions, has not been evident from clinical trials, possibly because of the fact that cancer patients with advanced disease have impaired ability to form antitherapeutic antibodies. The roles of intrinsic antibody effector functions and Fc receptor binding in ADC activity have also not been established definitively. The 3 ADCs that achieved FDA approval had very different antibody characteristics: GO (IgG4 subtype) has no effector function activity,3 brentuximab vedotin (IgG1 subtype) has modest antibody-dependent cellular phagocytosis only,23 and ado-trastuzumab (IgG1 subtype) emtansine is highly active in effector function assays.24
Most ADCs that are currently in clinical development are either humanized or fully human.17 Although there is a mixture of ADCs with intact or abrogated antibody-dependent cellular cytotoxicity, none has been specifically engineered to enhance effector function. There has been much investigation of the potential impact of smaller antibody fragments such as diabodies or minibodies on ADC activity.18,25 These molecules generally demonstrate enhanced tumor penetration but more rapid clearance. Although these characteristics have advantages for radiolabeled imaging reagents, it is not clear that they would result in better drug delivery, specificity, or antitumor activity.25 An extension of the ADC concept is the use of small-molecule delivery vehicles. These molecules will have significant pharmacokinetic differences compared with ADCs, including rapid clearance and larger volume of distribution, as well as a lack of immune effector functions. An example of this approach is vintafolide, which uses folic acid to deliver a vinblastine analog to cells expressing the folate receptor.26 The relative advantages of one approach over the other may be difficult to interpret unless the same target, cytotoxic agent, and clinical indication are compared directly.
Chemical conjugation
Traditionally, drugs have been attached to antibodies using native amino acids, taking advantage of the chemical reactivity of the ε-amino terminus of lysine residues or the sulfhydryl portion of a reduced cysteine residue.27 When using the lysine residues, the stoichiometry and location of the resulting adducts represents a distribution across many of the lysine residues in the antibody. In contrast, conjugation to reduced cysteines results in even-numbered drug additions (ie, 2, 4, 6, or 8), depending on how many of the interchain disulfide bonds are reduced. These additions occur in the hinge region of the antibody and have no impact on antigen binding and little impact on binding to Fc receptors on immune cells. To date, most ADCs have targeted an average of 4 drugs per antibody. This ratio was chosen as an optimal combination of cytotoxicity and pharmacokinetic stability.11,28,29
In recent years, significant effort has been devoted to developing site-specific conjugation methods either by introducing novel unpaired cysteine residues or nonnatural amino acids that allow for orthogonal chemistry methods.30-32 The impact of site-specific conjugation methods has not yet been evaluated in clinical trials.
Linker technology and drug release
An ADC linker should function as a highly stable bridge between the antibody and payload that allows efficient drug release upon delivery inside malignant cells. Early efforts to conjugate cytotoxic agents to antibodies involved conditionally stable linkers, including hydrazones that undergo cleavage under low pH conditions, such as is present in lysosomes. GO was the first such ADC to demonstrate significant clinical benefit, targeting the CD33 antigen expressed on normal and malignant myeloid cells. The hydrazone linker in this drug was shown to be unstable,33 which may have compromised overall activity and contributed to nontarget toxicity. Greater stability has been obtained with cleavable peptides in combination with self-immolative groups that undergo intracellular enzymatic degradation. The most advanced example incorporating such a linker is brentuximab vedotin, in which a dipeptide substrate for the lysosomal protease cathepsin B results in drug release.11 Finally, several ADCs, including the recently approved ado-trastuzumab emtansine, have linkers that release modified drugs through antibody degradation.12 Increased plasma stability and efficient intracellular drug release continues to be a focus for ADC research.
Cytotoxic agents
The released drug or “payload” of an ADC must be present in sufficient concentration inside the tumor cells to cause cell death. Early attempts at ADCs included toxins such as ricin-A chain and Pseudomonas exotoxin.34 These molecules posed challenges due to immunogenicity. Subsequently, chemotherapy drugs such as Adriamycin were used on ADCs.20 However, these molecules proved to have insufficient single-agent activity to justify further development. In the last 2 decades, it has become apparent that highly potent cytotoxic agents ranging from 100-10 000 times more potent than standard chemotherapy agents were needed. As discussed previously, drug release commonly occurs in the lysosome due to proteolytic cleavage or acid hydrolysis. However, the drug exerts its cell-killing activity elsewhere, such as the cytoplasm or nucleus. Identifying cytotoxic agents that are resistant to lysosomal proteases and are able to exit the lysosome (via diffusion or facilitated transport) are additional considerations for the design of ADCs.
GO used calicheamicin, a DNA-damaging agent with highly potent cytotoxicity, as the payload.35 Systemic administration of the free drug is not feasible due to toxicity, but by directing the drug to tumor cells in an ADC format, much lower systemic doses can achieve high concentrations in tumor cells. The 2 recently approved ADCs, brentuximab vedotin11 and ado-trastuzumab emtansine,12 each use a highly potent microtubule-disrupting agent, monomethyl auristatin E (a synthetic analog of dolastatin 10) and DM1, a maytansine derivative, respectively. This class of cytotoxic agents has an additional level of specificity in that it preferentially kills proliferating cells by disrupting the mitotic spindle apparatus. Furthermore, malignant cells frequently have disrupted cell cycle checkpoints, increasing the sensitivity to mitotic catastrophe compared with healthy cells.
ADCs may be subject to many of the same potential mechanisms of resistance as standard chemotherapy agents.36 In addition, a potential mechanism of resistance to ADC therapy is the possible loss of expression of the molecular target or down-modulation such that the target is no longer accessible on the cell surface. The extent to which each of these mechanisms of resistance affects the use of ADCs is currently being investigated. It is likely that the future of ADCs will bring a diversity of payloads and new mechanisms of action, some of which may be able to overcome acquired resistance.
Present and future of ADCs
Today, there are 2 ADCs available for patients in the United States. However, with more than 30 additional molecules in clinical trials (Table 2),19 it is likely that number of approved ADCs will increase substantially in the coming decade. Moreover, this class of drugs provides a new opportunity to re-examine the future of cytotoxic therapy. The combination of increased potency with better tolerability offers the hope for curing more cancers and, for those cancers that cannot be totally eradicated, extended therapy and an improved quality of life for patients. A century after Paul Ehrlich, his challenge is being taken up by a new generation of scientists who are working diligently to improve the specificity and activity of cancer chemotherapy. Although ADCs have just recently come of age, the evolution of the field is rapidly accelerating and the impact on cancer care is likely to be great.
Disclosures
Conflict-of-interest disclosure: The authors are employed by and have equity ownership in Seattle Genetics. Off-label drug use: Data are presented from clinical trials of antibody-drug conjugates that have not been approved.
Correspondence
Jonathan Drachman, Seattle Genetics Inc, 21823 30th Dr SE, Bothell, WA 98021; Phone: 206-598-3300; Fax: 425-527-4315; e-mail: jdrachman@seagen.com.
