
Abstract
Nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) is a unique diagnostic entity, with only ∼ 500 new cases in the United States per year with a similar infrequent incidence worldwide. NLPHL also has distinctive pathobiology and clinical characteristics compared with the more common classical Hodgkin lymphoma (cHL), including CD20 positivity of the pathognomic lymphocytic and histiocytic cells and an overall more indolent course with a higher likelihood of delayed relapses. Given the limited numbers of prospective NLPHL-focused trials, management algorithms historically have typically been centered on retrospective data with guidelines often adopted from cHL and indolent B-cell lymphoma treatment approaches. Key recent publications have delineated that NLPHL has a higher level of pathological overlap with cHL and the aggressive B-cell lymphomas than with indolent B-cell lymphomas. Over the past decade, there has been a series of NLPHL publications that evaluated the role of rituximab in the frontline and relapsed setting, described the relative incidence of transformation to aggressive B-cell lymphomas, weighed the benefit of addition of chemotherapy to radiation treatment for patients with early-stage disease, considered what should be the preferred chemotherapy regimen for advanced-stage disease, and even assessed the potential role of autologous stem cell transplantation for the management of relapsed disease. General themes within the consensus guidelines include the role for radiation treatment as a monotherapy for early-stage disease, the value of large B-cell lymphoma–directed regimens for transformed disease, the utility of rituximab for treatment of relapsed disease, and, in the pediatric setting, the role of surgical management alone for patients with early-stage disease.
Introduction
Hodgkin lymphoma (HL) remains a rare hematologic malignancy worldwide, with a predicted 9290 new diagnoses in the United States in 2013.1 The majority of these HL diagnoses will be classical HL (cHL), with nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) comprising only ∼ 5% of these HL cases. This pathologic entity was first described in 1944 by Jackson and Parker as a paragranuloma histologic variant of HL.2,3 This diagnosis was referred to by several alternate terms until 1994, when the Revised European-American Lymphoid (REAL) malignancy classification characterized the lymphocyte-predominant subtype of HL by the presence of the distinct CD20-positive lymphocytic and histiocytic (L&H) cells. The disease was then later renamed by the World Health Organization (WHO) in 2001 as NLPHL, which remains the diagnostic term used today.4
Evolution of understanding of pathogenesis
Given the typical clinical behavior pattern of NLPHL as an indolent lymphoma, as well as the characteristic CD20 positivity of L&H cells (Figure 1) with a lack of CD30 or CD15 positivity seen in cHL, it was long commonly viewed that NLPHL likely would have the closest pathogenesis overlap with indolent B-cell non-Hodgkin lymphoma diagnoses such as follicular lymphoma.5 A complicating factor in analysis was that, similar to Hodgkin and Reed/Sternberg (HRS) cells in cHL, in NLPHL, the L&H cells were rare within the tumor mass, making up < 1% of the overall tumor cells. It was known that both HRS and L&H cells arise from germinal center B cells, but determining the key differences in CD20, CD30, and CD15 expression was often challenging. We now know that, compared with HRS cells, L&H cells express IgV genes, BCL6, and activation-induced cytidine deaminase; however, CD10 and CD19 expression are typically absent.6-9 L&H cells also generally grow within expanded B-cell follicles and are often encircled by germinal-center T-helper cells that are positive for CD4 and CD57.10

NLPHL key pathologic features. (A) Magnification of 20× showing the nodular formation. (B) Magnification of 400× with staining for CD20 highlighting L&H cells in a background of reactive lymphocytes and histiocytes. Images courtesy of Jeffrey Medeiros, MD, University of Texas MD Anderson Cancer Center.
NLPHL key pathologic features. (A) Magnification of 20× showing the nodular formation. (B) Magnification of 400× with staining for CD20 highlighting L&H cells in a background of reactive lymphocytes and histiocytes. Images courtesy of Jeffrey Medeiros, MD, University of Texas MD Anderson Cancer Center.
The work of Brune et al significantly enriched our understanding of the pathogenesis of NLPHL by establishing a method of microdissection of L&H cells from frozen tissue, which allowed for RNA isolation from these cells and further global gene expression profiling analysis.11 Unsupervised hierarchical clustering, followed by principal component analysis of the mean expression vectors for each diagnostic entity, showed that NLPHL was most closely related to T-cell–rich B-cell lymphoma (TCR-BCL), a subset of diffuse large B-cell lymphoma (DLBCL), and cHL, despite having differences in immunohistochemical expression from these diagnoses. Contrary to previously held viewpoints, NLPHL was not found to have substantial gene expression profiling overlap with follicular lymphoma. Supervised analysis confirmed these results and 49 distinctive genes were identified as being reliably up-regulated in L&H cells, allowing for the generation of a comparative profile of NLPHL compared with cHL, normal B cells, and indolent B-cell non-Hodgkin lymphoma. L&H cells had overlap with germinal center B cells as the point of transition to memory B cells. The assessment of the function of the genes differentially expressed in L&H cells also demonstrated that they work to produce an immunosuppressive environment. L&H cells also showed high-level expression of NF-κB activity and ERK activation. These insights into the pathogenesis of NLPHL opened up future methods that could be used for molecular diagnosis and therapeutic development and highlighted that, although there can be clinical behavior overlap between 2 lymphoma diagnoses, their underlying driving biologic factors for growth can be very divergent.
Additional distinguishing pathologic features of NLPHL have also been described. SOCS1, which is known to control JAK2 activity, has been found to undergo somatic hypermutation (SHM) in NLPHL, which can result in the activation of JAK2 in nearly 40% of NLPHL cases via activation of the JAK2/STAT6 pathway.12 STAT6 is also constitutively activated in cHL. The mechanism leading to high NF-κB activity in NLPHL has also been found to be different from that in cHL. In cHL, inactivating mutations occur commonly in NFKBIA and TNFAIP3, which code for negative NF-κB regulators Iκβα and A20, whereas in NLPHL, there is a low mutation frequency.13 SHM has also been shown to be similar to DLBCL and cHL in contributing to the molecular pathogenesis of NLPHL. In a small study, 80% of NLPHLs had mutations in 1 or more of SHM targets, with PAX5 being the most frequently affected.14 A CD4+CD8+ T-cell subset has also been described in 58% of NLPHL cases and its presence might represent an activated or reactive T-cell population. In addition, because this population of T cells was rarely seen in cHL or reactive lymphoid hyperplasia without progressively transformed germinal centers, the presence of CD4+CD8+ T cells might be useful in supporting a diagnosis of NLPHL in cases in which there is limited tissue.15
Potential role of familial risk
In 2004, the potential for familial association for NLPHL was first described in siblings and now a total of 4 familial-associated cases have been reported.16-19 A germline candidate mutation in nuclear protein ataxia-telangiectasia (NPAT) was also described in a Finnish family with NLPHL.20 This year, a Finnish Cancer Registry–based review of data was reported and involved data collection of first-degree relatives from nearly 700 patients with NLPHL.21 The standardized incidence ratio for NLPHL in the first-degree relatives was 19, which was higher compared with cHL at 5.3. This suggests that potential as-yet unknown genetic or environmental factors are influencing this familial incidence ratio and potentially could be used to further develop and select therapies through improved knowledge of the disease process.
Natural disease history and therapeutic strategies
The rarity of the diagnosis of NLPHL makes it very challenging to accrue the numbers of patients needed to complete the same types of large randomized trials that are done in cHL. Therefore, most of the data that support management decisions comes from single-arm phase 2 and retrospective trials. Furthermore, overall NLPHL compared with HL is generally characterized as having a more indolent course with delayed relapses. The German Hodgkin Study Group (GHSG) conducted a large, retrospective study of nearly 400 patients and, as anticipated, the majority 79% had early-stage disease. With a median follow-up of 50 months, the freedom from treatment failure (FFTF) and overall survival (OS) was better for NLPHL compared with cHL at, respectively, 88% versus 82% and 96% versus 92%, with early-stage patients having better outcomes compared with those with advanced-stage disease.22 Improved outcomes for FFTF for early-stage patients compared with advanced-stage patients were also reported by the European Task Force on Lymphoma, with 85% for stage I, 71% for stage II, 62% for stage III, and 24% for stage IV.23 It is also hoped that advances in our knowledge of the pathogenic processes that drive NLPHL will open up further development of biologically targeted therapeutic options, because patients with relapsed NLPHL have historically been excluded from most phase 1 novel agent clinical trials.
Early-stage disease
Although radiation as a single modality for treatment would be considered inferior treatment for patients with early-stage cHL, multiple trials describe excellent outcomes for radiation treatment alone, which has led to its being accepted as the standard of care management approach for NLPHL patients with stage IA or IIA disease. More recent data from the British Columbia Cancer Agency (BCCA) suggests potentially improved outcomes for combined modality (CM) treatment with brief courses of ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) followed by involved field radiation therapy (IFRT). CM treatment is typically considered for patients with stage IB or IIB disease, although the preferred regimen of chemotherapy remains debatable given the lack of comparative trials.24 In addition, recent trials have also demonstrated a positive benefit for the CD20-targeted monoclonal antibody rituximab, which was initially evaluated in relapsed NLPHL disease for patients with stage IA disease. Pediatric trials have shown excellent results from surgical excisional biopsy-alone management for children with stage IA disease (Table 1).25

Radiation therapy.
Multiple large, cooperative groups such as the Australasian Radiation Oncology Lymphoma Group and the GHSG, as well as smaller single-institution series including the Harvard Hospital group and our center, The University of Texas MD Anderson Cancer Center (UTMDACC), have reported excellent outcomes from radiation therapy (RT) alone. The Australasian group described the potential of RT alone to cure NLPHL stage I and II patients and delineated outcomes for patients treated from 1969 through 1995. Overall, 80% of patients had stage I disease. Given the time period of this retrospective study, 52% of patients received full mantle field irradiation. At 15 years of follow-up, the freedom from progression (FFP) was 84% in stage I and 73% in stage II patients.26 Researchers from UTMDACC demonstrated similar positive benefit for stage IA and IIA patients treated during an overlapping time period. Limited-field RT (LFRT) was delivered to 78% of the patients with a median dose of 40 Gy. With a median follow-up of 8.8 years, only 1 of the LFRT patients experienced disease relapse and no secondary malignancies were seen in this patient group. The best outcomes were described in stage IA patients, who had a 5-year relapse-free survival of 95%.27
Most recently, in 2010 Chen et al reported on long-term outcomes for 113 early-stage patients treated at the Harvard-based Joint Center for Radiation Therapy. This study evaluated a large number of patients relative to the incidence of this diagnosis and had a long median follow-up of > 11 years. A total of 93 patients received RT alone, with 27% receiving LFRT, defined as IFRT or less than mantle or mini-mantle RT. Ten-year progression-free survival (PFS) was 85% for stage I and 61% for stage II with OS ratesof 94% and 97%, respectively. PFS and OS were not statistically different for those treated with LFRT, regional-field, or EFRT. However, by comparison, 86% of the patients treated with chemotherapy alone had relapse of disease.28 Further data supporting IFRT for management of stage IA disease comes from the GHSG, who analyzed outcomes for 131 patients treated across their HD4, HD7, HD10, and LPHL IA trials and compared IFRT, EFRT, and CM treatment options. Complete remissions (CRs) occurred in 98% of patients and outcomes were equivalent across the 3 treatment arms with the exception of patients included from the HD7 trial. In this 2-arm randomized trial, patients treated with 2 cycles of ABVD followed by EFRT plus IFRT had superior 7-year FFTF compared with those treated with EFRT plus IFRT alone, at 96% versus 83%, respectively.29
CM therapy and chemotherapy alone.
The potential benefit of adding nonanthracycline chemotherapy to RT for early-stage patients was explored at UTMDACC from 1963 to 1996, when stage I or II very favorable or favorable patients based on definitions from the European Organization for Research and Treatment of Cancer (EORTC) and Groupe d'Etude des Lymphomes de l'Adulte (GELA) were treated with RT alone versus CM therapy. The 23% of patients who received the CM approach were treated with a median of 3 cycles of mechlorethamine, vincristine, procarbazine, and prednisone (MOPP) or with mitoxantrone, vincristine, vinblastine, and prednisone (NOVP). A median dose of 40 Gy was given to both treatment groups. Overall, MOPP or NOVP chemotherapy did not improve relapse-free survival, with similar outcomes at 10 years of 77% and 68% for RT alone and CM treatments, respectively.30 Nonanthracycline chemotherapy has also been evaluated in children and adolescents with early-stage disease as a method of minimizing intensity of treatment and thus adverse events in this young patient population. From 2004 to 2006, 18 patients with stage IA or IIA disease received 3 cycles of cyclophosphamide, vincristine, prednisolone (CVP) with a CR rate of 94%. At a short follow-up of 12 months, the event-free survival was 94%.31
The potential benefit of short-course anthracycline-based chemotherapy was described by the Groupe Ouest-Est d'Etude des Leucèmies et Autres Maladies du Sang (GOEL-AMS). Patients within their H81 and H90 trials received ABVD or epirubicin, bleomycin, vinblastine, methotrexate (EBVM) for 1 or 3 cycles followed by RT. Of the 500 stage IA and IIA HL patients, 8% had NLPHL. Overall outcomes, including 15-year mortality rates, were very low at < 2.5% across both the cHL and NLPHL patients without mediastinal adenopathy; however, it is difficult to conclude superiority for CM over RT alone treatment in NLPHL patients from this study.32 More recently, the BCCA found that treating early-stage NLPHL patients with ABVD improved PFS.24 Within this retrospective trial, stage IA/B or IIA patients who received RT alone were compared with those who received ABVD for 2 cycles followed by RT. More than 60% had stage I disease and the 10-year PFS showed favorable outcomes for CM treatment at 91% versus 65% and raised the question of whether all early-stage HL patients should be treated similarly.
Rituximab monotherapy.
Given the high levels of activity seen for the anti-CD20 antibody rituximab from trials conducted in the relapsed setting, with overall response rates ranging from 93% to 100% in the GHSG and Stanford trials, respectively, a phase 2 GHSG study was undertaken in the frontline setting for patients with stage IA disease.33,34 Four weekly treatments were given, with an overall response rate (ORR) of 100% and a CR rate of 86% in the 28 patients evaluated; the 3-year PFS was 81%, suggesting that long-term results remain superior with CM or RT, but opening further potential for rituximab-based combination treatments in the frontline setting.35 Additional outcomes data for frontline monotherapy with rituximab comes from the Stanford group, which treated newly diagnosed patients with initial 4 weekly infusions rituximab at a dose of 375 mg/m2 with potential for maintenance therapy every 6 months for 2 years. In total, 19 newly diagnosed patients have been treated and 68% have stage I/II disease. Ten patients received limited treatment and 9 had treatment that included maintenance therapy. The ORR was 100% with a CR/unconfirmed CR (CRu) rate of 63%. Late relapses were seen and at 5 years the estimated PFS is 52% while at 10 years it is 35% with no significant difference noted for those who did or did not receive maintenance therapy. The median PFS for stage I/II patients was 5.6 years. Ten patients overall had relapse with 60% having biopsy-documented transformation to DLBCL or TCR-BCL. Four of the 6 patients with transformation had stage III disease with abdominal involvement at time of diagnosis.36
Surgical resection alone.
The overwhelming majority of the data for this approach comes from the pediatric setting, where it is especially desired to minimize exposure to RT and/or chemotherapy. Two initial reports were published for 19 children treated with surgical resection alone in 2003, highlighting the potential benefits of this approach.25,37 Subsequent multicenter trials have confirmed the benefit of this approach. The EuroNet-Paediatric Hodgkin Lymphoma Group (PHL) evaluated outcomes for 57 pediatric patients. CR was achieved after surgery in 86% of patients, all with stage IA disease. The relapse rate was 27% in the CR group, with all relapses occurring early (within 26 months) and 56% being local. At a median follow-up of 43 months, the estimated overall FFP was 67%.38 Additional support for this approach comes from the Children's Oncology Group (COG), who treated a total of 52 patients with stage IA disease without bulky disease with surgical total resection alone. These investigators found a 17% relapse rate at a median of 10 months, with a 2-year estimated event-free survival of 80% and OS of 100% at a median follow-up of 26 months. Although follow-up overall for both of these trials was relatively short, if these results remain durable, with longer follow-up, this would add further evidence to this as a potential curable treatment modality.39
Advanced-stage disease
Given that at least 70% to 80% of patients with NLPHL are diagnosed with early-stage disease, defining the optimal treatment regimen for advanced-stage disease is even more challenging. The rarity of the diagnosis of advanced-stage NLPHL generally restricts the ability to perform large comparative trials, so the best evidence for determining preferred management options typically comes from retrospective comprehensive analysis of mainly cHL enrolled treatment trials or single institution published experiences. For this reason, treatment guidelines such as those from the National Comprehensive Cancer Network list multiple chemotherapy options such as CVP, CHOP (cyclophosphamide, hydroxydaunorubicin, vincristine, prednisone/prednisolone), and ABVD plus or minus rituximab as valid options (Table 2).
Comparative outcomes for advanced-stage NLPHL treatments

N/R indicates not reported; COPP, cyclophosphamide, vincristine, procarbazine, prednisone; IMEP, ifosfamide, methotrexate, etoposide, prednisone; and BEACOPP, bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone.
cHL-directed chemotherapy.
The GHSG analyzed outcomes for the 8298 HL patients treated on their HD4 to HD12 trials. Of the patients enrolled, only 5% had NLPHL. CR rates for frontline treatment for advanced-stage NLPHL patients mirrored those seen for the cHL patients at 77%. Although at a median follow-up of 50 months, the FFTF for advanced-stage NLPHL and cHL was similar at 77%, there was a higher rate of relapses at 1 year or greater for those with NLPHL, at 7.4% versus 4.7%.22
Data have also been presented from both the Cancer and Leukemia Group B (CALGB) trials 8251 and 8952 and the Dana-Farber Cancer Institute and Joint Center for Radiation Therapy describing comparative outcomes from patients treated with ABVD or etoposide, vinblastine, and doxorubicin (EVA) compared with the alkylating agent–containing regimen mechlorethamine, vincristine, procarbazine, prednisone (MOPP) or the hybrid regimen MOPP/ABVD. Of the 37 NLPHL patients treated across these trials, there was a 2-fold lower rate of disease progression or relapse in those patients treated with MOPP or MOPP/ABVD compared with ABVD. These investigators proposed that alkylating agent–based regimens such as MOPP or cyclophosphamide-based therapies might be preferred.40,41
B-cell lymphoma–directed chemotherapy.
We at UTMDACC have conducted a retrospective analysis of the B-cell lymphoma regimen R-CHOP in patients with NLPHL from 1995 to 2010. Of 83 patients referred with NLPHL, we confirmed NLPHL diagnoses in 63 patients. Advanced-stage disease was documented in 24 patients, who received treatment regimens including mitoxantrone, vincristine, vinblastine, prednisone (NOVP), ABVD plus or minus rituximab, and R-CHOP. Twelve advanced-stage patients received R-CHOP. ORR with R-CHOP was 100%, with a 90% CR rate, and no relapses or transformations have been seen at a median follow-up of 42 months.42
Relapsed and transformed disease
Immunotherapy for relapsed disease.
Given the known near 100% level of CD20 expression on L&H cells, 2 key trials by the GHSG and Stanford first evaluated the role of rituximab in treating relapsed NLPHL disease.34 The Stanford phase 2 trial initially treated a total of 23 NLPHL patients, 11 patients of whom had previously untreated disease. Patients were treated with 4 weekly doses at 375 mg/m2. All patients entered into remission, with a CR/CRu rate overall of 56%. This trial was then modified to allow for extended maintenance rituximab treatment, with patients repeating the 4 weekly infusions every 6 months for 2 years. A total of 16 patients, 7 with relapsed disease, received extended therapy. CR/CRu increased to 88%. No significant differences were noted in remission rates for newly diagnosed compared with previously treated patients. Progression of disease was lower in extended treatment, occurring in 2 patients compared with 15 patients with limited treatment. Median FFP was 24 months for limited therapy and was not reached for extended therapy. Across limited and extended treatment, 4 patients had transformation to DLBCL, with 1 patient dying of this disease.43
The GHSG conducted a similar trial of limited rituximab therapy for NLPHL patients with relapsed or refractory disease. A total of 21 patients were treated and, with central pathology review, the diagnosis of NLPHL was confirmed in 15 patients; 2 were reclassified as TCR-BCL and 4 as CD20+ cHL. In those with confirmed NLPHL, the ORR was 94%, with 53% CRs. At a median follow-up of 63 months, the median time to progression was 33 months (Table 3).33
Limited versus extended rituximab treatment for relapsed NLPHL

TTP indicates time to progression.
A tositumomab and 131I tositumomab phase 1/2 trial has also been conducted for patients with relapsed HL. Seven total patients were treated, with all but 1 patient having cHL. Patients were treated with single-patient-specific doses of 450 mg of tositumomab and 131I tositumomab. The patient with NLPHL did enter into CR. Overall, the most common adverse events, as anticipated, were cytopenias.44
Transformation to aggressive B-cell lymphoma.
Overall, a higher risk of transformation to aggressive B-cell NHL exists for NLPHL than for cHL. One of the largest series was published by the BCCA group.45 Of the 95 patients followed, 14% experienced transformation to an aggressive B-cell NHL at a median of 8.1 years, with an actuarial risk of transformation at 20 years of 30%. An increased risk of transformation was seen in those with advanced-stage disease and splenic involvement at diagnosis of NLPHL. Of the 13 patients with transformation, 6 patients received R-CHOP chemotherapy, with 6 patients receiving R-CHOP or other rituximab-containing chemotherapy regimens, followed by high-dose chemotherapy and autologous stem cell transplantation (ASCT). CR in those with transformation was achieved in 69% and the 10-year PFS and OS rates were 52% and 62%, respectively.
The Mayo Clinic identified 222 patients with NLPHL who were seen over a similar 40-year time period.46 Compared with the BCCA outcomes, patients from the Mayo series had a lower rate of transformation at 8%, with transformation occurring earlier and a median time to transformation of 2.9 years. Of the 17 patients with transformation, 9 were treated with R-CHOP or CHOP chemotherapy and 2 patients went on to ASCT. Overall outcomes were favorable, with a 5-year OS for the transformed lymphoma patients of 76%.
Although the clinical factor of advanced-stage disease at diagnosis is a risk factor for transformation, the pathological risk factors for transformation remain under investigation. A group from Saudi Arabia has found that a T-cell-rich microenvironment around the L&H cells compared with a B-cell-rich background is associated with a higher rate of transformation, with 17% of the T-cell-rich cases having transformation to aggressive lymphomas, although OS was similar.47
ASCT.
Studies evaluating the role of ASCT in relapsed NLPHL and transformed disease is limited. Jackson et al defined outcomes for a series of 8 patients who underwent ASCT within an 88 NLPHL patient retrospective study. They described an overall median failure-free survival of 39.2 months.48 The French Adult Study Lymphoma Group evaluated 106 patients with NLPHL. Of these, 66 patients experienced transformation at a median of 4.7 years. A total of 19 patients with transformation underwent curative intent therapy, with 9 patients undergoing ASCT and 10 patients receiving other types of chemotherapy. OS was similar between these 2 groups, with a 10-year OS rate after transformation of 60%.49 Outcomes for relapsed HL patients treated with ASCT have also been compared for 299 cHL versus 19 NLPHL patients, with similar 5-year PFS and OS of 50% compared with 39% and 56% compared with 53%, respectively.50
Karuturi et al from UTMDACC recently published on results for NLPHL management with ASCT. In total, 26 patients underwent high-dose chemotherapy and ASCT, with 70% having NLPHL at time of transplantation and 30% having transformation to DLBCL. The 5-year PFS and OS for those with NLPHL were 61% and 73%, respectively, compared with 87% and 87%, respectively, for DLBCL patients.51 These small retrospective trials suggest that ASCT is a reasonable approach for the management of patients with relapsed disease, particularly those with multiply relapsed NLPHL or transformation to DLBCL.
Conclusions
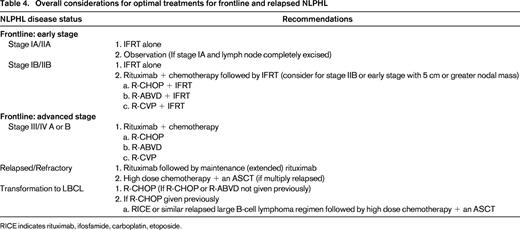
Clear progress has been made in delineation of the driving pathologic features of NLPHL and in defining best management approaches (Table 4). However, to solidify what should be the preferred therapy for each presenting stage group, an increased focus on randomized clinical trials is needed. Given the rarity of this diagnosis, these trials would need to be intergroup collaborative protocols. Without this type of approach, it will not be feasible for us to collectively determine whether CM treatment in early-stage disease has significant added benefit compared with RT alone, nor will we be able to decide what should be the best chemotherapy regimen for patients with advanced-stage disease. It is also of key importance that NLPHL patients are included in phase 1 and 2 novel therapeutic trials or treatment paradigms for these patients will not continue to evolve compared with patients with cHL or B-cell NHL. Similar to the highly positive outcomes with rituximab, new agents could have the potential of providing significant benefit while hopefully having relatively low toxicity levels and preserved quality of life. This is of particular importance given the natural clinical history of this diagnosis.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Off-label use of rituximab discussed.
Correpondence
Michelle Fanale, University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Unit 429, Houston, TX 77030; Phone: 713-792-2860; Fax: 713-794-5656; e-mail: mfanale@mdanderson.org.
