
Abstract
MYC is a potent oncogene initially identified as the target of the t(8;14)(q24;q32) chromosome translocation in Burkitt lymphoma. MYC gene alterations have been identified in other mature B-cell neoplasms that are usually associated with an aggressive clinical behavior. Most of these tumors originate in cells that do not normally express MYC protein. The oncogenic events leading to MYC up-regulation seem to overcome the inhibitory effect of physiological repressors such as BCL6 or BLIMP1. Aggressive lymphomas frequently carry additional oncogenic alterations that cooperate with MYC dysregulation, likely counteracting its proapoptotic function. The development of FISH probes and new reliable antibodies have facilitated the study of MYC gene alterations and protein expression in large series of patients, providing new clinical and biological perspectives regarding MYC dysregulation in aggressive lymphomas. MYC gene alterations in large B-cell lymphomas are frequently associated with BCL2 or BCL6 translocations conferring a very aggressive behavior. Conversely, MYC protein up-regulation may occur in tumors without apparent gene alterations, and its association with BCL2 overexpression also confers a poor prognosis. In this review, we integrate all of this new information and discuss perspectives, challenges, and open questions for the diagnosis and management of patients with MYC-driven aggressive B-cell lymphomas.
Introduction
MYC was initially identified as the target oncogene dysregulated by the t(8;14)(q24;q32) translocation in Burkitt lymphoma (BL). MYC rearrangements involving the heavy- and light-chain immunoglobulin (IGL) loci and different non-IG genes were subsequently detected in other lymphoid neoplasms usually associated with very aggressive clinical behavior.1,2 The transforming oncogenic potential of MYC was initially demonstrated in cell lines and transgenic animal models.3 However, MYC dysregulation alone does not cause lymphoma,4 and the t(8;14) has been found at very low levels in blood and BM of healthy individuals,5 indicating that this genetic alteration per se is not sufficient to trigger lymphomagenesis. BL and most lymphomas carrying MYC translocations are among the most proliferative tumors. However, MYC expression has been difficult to identify in the germinal center (GC), the lymphoid cell compartment with the highest proliferative fraction, where most of these tumors originate. Understanding the possible role of MYC in normal lymphoid regulation and particularly the modulation of the GC reaction has been elusive.
Recent studies, including basic immunology analyses, new animal models, next-generation sequencing, and clinicopathological observations, are converging to provide a new perspective of the role of MYC in the lymphoid system and the pathogenesis of aggressive lymphomas. In this review, we integrate all of this new information and discuss new perspectives, challenges, and open questions for the diagnosis and management of patients.
MYC as a transcription factor
MYC is a transcription factor forming heterodimers with the related protein MAX that bind to promoter regions of target genes and modulate their expression by the recruitment of specific coactivators and repressors.1-3 Transcriptional activation of MYC is mediated by binding of the histone acetyltransferases CBP/p300 and TIP60/GCN5 or the transcription factor P-TEFb, among others. Transcriptional repression is modulated by several mechanisms, including the interaction of these complexes with the transcription factor MIZ-1, preventing recruitment of the activating molecule p300 and facilitating binding of the gene silencing DNA-methyltransferase DNMT3a. Other transcription factors, such as MAD, may titrate out MYC from the complexes and then the MAX/MAD heterodimers recruit histone deacetylases (HDACs) that repress gene transcription.3 MYC may also repress gene expression by recruitment of HDAC to promoters containing E-boxes.6
The transcriptional program regulated by MYC includes 10% to 15% of all human genes. The main cell functions and pathways under control of MYC are cell proliferation and growth, DNA replication, protein biosynthesis, and regulation of metabolism and energy. MYC promotes the transition from the G0/1 phase to the S phase, activating directly and indirectly the expression of CCND2 and CDKs and down-regulating cell cycle inhibitors.1,2,7,8 The MYC transcriptional network also includes the direct regulation of a large number of miRs with oncogenic or tumor suppressor function.9,10 MYC up-regulates the oncogenic miR 17-92 cluster, but most miRs directly regulated by MYC are usually repressed.9 The miR17-92 polycistron at 13q31 is commonly amplified in several subtypes of aggressive lymphomas11,12 and its oncogenic function is mediated in part by the down-regulation of PTEN, TP53, and E2F1, facilitating the activation of the PI3K/AKT pathway and inhibition of apoptosis, respectively.3 MYC represses several miRs with tumor suppressor function by the recruitment of HDACs.13 These miRs include miR15a/16-1, miR26a, miR29, and miR34, which regulate crucial functions in neoplastic development such as apoptosis (miR15a/16-1 and miR34 targeting BCL2 and TP53, respectively), proliferation (miR29a targeting CDK6), or cell differentiation (miR26a targeting EZH2).3,14,15 MYC itself is also negatively regulated by some miRs, such as miR34 and miR494.14,15 miR494 is in turn repressed by EZH2, creating a positive autoregulatory loop (MYC/miR26a/EZH2/miR494) that sustains the persistent expression of MYC and EZH2, promoting the malignant phenotype of cells.14 The interactions of the networks regulated by MYC and its target miRs are complex and suggest fine-tuning of different processes that may be targeted by new therapies.13,16
Intriguingly, the gene profile transcriptionally regulated by MYC varies in different cell types with relatively little overlap.17 Two recent studies shed light on this puzzling observation, showing that, instead of activating a particular gene signature, MYC acts as an amplifier of the transcribed genes in a given cell by uploading to the promoters of active genes and enhancing their transcription.18,19 MYC does not bind to promoters of silent genes and therefore acts as an activator of the preexisting transcription program. This function of MYC may be relevant to understanding the increased aggressiveness of tumors associated with other oncogenic events carrying MYC alterations and may offer perspectives for new therapies.18,19
A paradoxical role of MYC is the induction of apoptosis.1,3 The biological meaning of this function is not fully understood. It has been interpreted as a protective mechanism of cells to counteract the effects of oncogenic activation and avoid propagation of transformed cells. The mechanisms of MYC-mediated apoptosis may involve several pathways. Overexpression of MYC increases DNA replication, possibly resulting in DNA damage that in turn triggers a TP53-mediated response, leading to apoptosis. MYC expression also seems to down-regulate, probably indirectly, antiapoptotic proteins such as BCL2 or BCLXL and up-regulate proapoptotic elements such as BIM.20 This antitumorigenic effect of MYC may explain in part the need of other cooperative mechanisms for cell transformation and tumor progression.
The relevant oncogenic role of MYC has stimulated the search for therapeutic strategies that may counteract its damaging functions. MYC protein itself has generally been considered “undruggable” and the potential approaches have been directed at reducing its expression, interfering with MAX dimerization or DNA binding, or acting on downstream target genes. However, most of these strategies have been difficult to apply in in vivo models.2,21,22 The recent discovery that MYC transcription depends on the regulatory function of BRD4 has offered new promising therapeutic opportunities.23,24 BRD4 is a member of the bromodomain and extraterminal (BET) subfamily of proteins that bind to lysine acetylated histones and recruit elements required for transcription. Two small molecules, JQ1 and iBET, displace BRD4 from acetylated chromatin, resulting in a down-regulation of MYC and modulation of its transcriptional program, including the up-regulation of MYC-repressed miRs, with a marked antiproliferative cell effect and tumor growth inhibition.16,23,24 These results have been confirmed in plasma cell myeloma (PCM) and BL cell lines with translocated IGH/MYC and also in aggressive lymphomas with MYC overexpression not related to structural gene alterations, suggesting that this strategy may be useful in a broad spectrum of MYC-driven tumors.16,23-25 Although BRD4 binds to a high number of enhancers and promoters, its inhibition is particularly sensitive in very large and active enhancers called super-enhancers that regulate oncogenes such as MYC. The addiction of PCM cells to MYC make the cells particularly sensitive to the BRD4-binding disruption on its super-enhancer.26
MYC regulation in GC cells
Most aggressive lymphomas with MYC alterations are related to follicular lymphoid cells, but the role of MYC in GC formation and maintenance has been elusive until recently.27,28 MYC is expressed in mature B cells initiating GC formation and in a small subpopulation of B cells of the light zone of the GC. However, MYC is absent in the highly proliferative cells of the GC dark zone. The sole expression of MYC in these selective subsets of B cells explains the failure of previous studies using bulk GC cells to detect its expression. In the early steps of GC formation, MYC is transiently up-regulated in few B cells before BCL6 is expressed (Figure 1). This expression seems to be induced by the initial interaction with antigens and T cells and is essential for GC formation because its abrogation results in a complete absence of GCs. In subsequent steps, BCL6 is up-regulated and directly represses MYC by binding to its promoter. This switch between MYC and BCL6 is associated with the formation of the dark zone of the CG and expansion of highly proliferating centroblasts. MYC is then reexpressed in a subset of activated cells of the light zone that have up-regulated NF-κB and express IRF4, whereas BCL6 is down-regulated. This MYC up-regulation is again dependent on antigen and T-cell interactions. The light zone MYC-positive cells seem to correspond to a selected subpopulation of B cells with high-affinity BCR that are prepared to reenter the dark zone for a subsequent round of proliferation and further acquisition of IG somatic mutations, perpetuating the GC reaction (Figure 1). MYC-negative cells in the light zone will probably be the subset primed to exit the GC as memory cells or early plasmablasts. BLIMP1 induction in these latter cells will promote the plasma cell differentiation program and will repress MYC expression by binding to its promoter (Figure 1).29

MYC expression and regulation in the formation of the normal GC reaction. MYC is initially expressed in B cells after interaction with antigens and T cells and is essential for GC formation. The subsequent up-regulation of BCL6 represses MYC and initiates the formation of the GC dark zone (DZ). MYC is reexpressed in a subset of cells of the light zone (LZ) after NF-κB up-regulation that will reenter into the DZ for subsequent rounds of IG somatic mutations. MYC-negative cells in the LZ exit the GC as memory cells or early plasmablasts. BLIMP1 induction will promote plasma cell differentiation and repress MYC.
MYC expression and regulation in the formation of the normal GC reaction. MYC is initially expressed in B cells after interaction with antigens and T cells and is essential for GC formation. The subsequent up-regulation of BCL6 represses MYC and initiates the formation of the GC dark zone (DZ). MYC is reexpressed in a subset of cells of the light zone (LZ) after NF-κB up-regulation that will reenter into the DZ for subsequent rounds of IG somatic mutations. MYC-negative cells in the LZ exit the GC as memory cells or early plasmablasts. BLIMP1 induction will promote plasma cell differentiation and repress MYC.
The absence of MYC in cells of the GC dark zone is intriguing and raises the question of the mechanisms sustaining the high proliferative program of these cells. Recent gene expression profiling studies of isolated cells from the GC dark and light zones have identified different transcriptional programs. TCF3 (E2A), a potent transcription factor highly expressed in the dark zone,30 up-regulates genes required for GC function, including CCND3 and E2F2, promoting cell proliferation.31 Interestingly, TCF3 induces its own negative inhibitor, ID3, that is also a target of MYC.32 These elements may create an autoregulatory loop controlling the transition of cells between the dark and light zones. ID3 expression promoted by TCF3 may contribute to the attenuation of the TCF3 program allowing the cell to move from the dark to the light zone (Figure 2A). The expression of MYC in light zone cells would sustain this effect by the induction of ID3.

Oncogenic mechanisms of MYC in aggressive mature B-cell lymphomas. (A) In BL and DLBCL, MYC is activated by gene translocations or amplifications. Activation of the TCF3/ID3 pathway cooperates with MYC in BL, whereas BCL2 and/or BCL6 translocations are the cooperating mechanisms in DLBCL. (B) In PBL, MYC is activated by translocations, whereas in ALK-positive large B-cell lymphomas, MYC is up-regulated indirectly by the oncogenic effect of ALK and STAT3 activation. In both tumors, MYC activation overcomes the suppressor effect of BLIMP1. The activation of the unfolded protein response may be a survival mechanism to counterbalance the proapoptotic function of MYC. Stars represent somatic mutations and green and red boxes indicate activating and suppressing mechanisms, respectively.
Oncogenic mechanisms of MYC in aggressive mature B-cell lymphomas. (A) In BL and DLBCL, MYC is activated by gene translocations or amplifications. Activation of the TCF3/ID3 pathway cooperates with MYC in BL, whereas BCL2 and/or BCL6 translocations are the cooperating mechanisms in DLBCL. (B) In PBL, MYC is activated by translocations, whereas in ALK-positive large B-cell lymphomas, MYC is up-regulated indirectly by the oncogenic effect of ALK and STAT3 activation. In both tumors, MYC activation overcomes the suppressor effect of BLIMP1. The activation of the unfolded protein response may be a survival mechanism to counterbalance the proapoptotic function of MYC. Stars represent somatic mutations and green and red boxes indicate activating and suppressing mechanisms, respectively.
MYC dysregulation in aggressive B-cell lymphomas
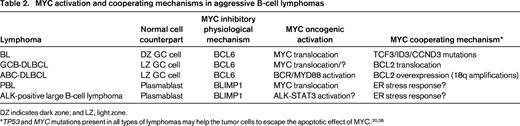
MYC gene alterations were initially identified in lymphoid neoplasms by cytogenetic and molecular genetic studies that recognized 8q24 translocations and MYC gene rearrangements, amplifications, or mutations.1 These studies were, however, difficult to apply on a routine basis in large series of patients. The development of MYC FISH probes and, more recently, a monoclonal antibody that specifically recognizes MYC protein in routinely processed tissues has simplified the analysis of these alterations in routine practice.33,34 These 2 technical advances have facilitated the study of MYC in large cohorts of patients, expanding our view and perspective of these alterations in different subtypes of aggressive lymphomas (Table 1). Intriguingly, most of these tumors originate in cells that do not express MYC protein. The oncogenic events leading to the up-regulation of MYC seem to overcome the inhibitory effect of physiological repressors such as BCL6 in GC cells or BLIMP1 in terminally differentiated B cells (Table 2). In addition, these aggressive lymphomas appear to have acquired additional oncogenic alterations that seem to cooperate with MYC dysregulation by counteracting especially its proapoptotic function (Table 2).

BL
BL is composed of highly proliferating mature B cells expressing a GC phenotype. It frequently presents in extranodal sites in children and young adults. Epidemiological studies have recognized 3 variants: endemic, sporadic, and HIV associated. The genetic hallmark of BL is the MYC translocation usually, with the IGH locus but also with IGL genes. These translocations are usually the sole chromosomal aberration or are associated with few additional alterations. In addition to MYC translocations, BL harbors also MYC and TP53 mutations in ∼ 60% and 40% of the cases, respectively.35,36 Similar MYC mutations have been identified in diffuse large B-cell lymphoma (DLBCL) and seem to be introduced via the GC somatic mutational machinery.37 Most of these mutations target functional domains that enhance the oncogenic potential of MYC by different mechanisms, including increased protein stability and transcriptional function, or by impairing the induction of the proapoptotic element BIM.37,38
Gene expression profiling studies have identified different signatures of BL and DLBCL.39,40 The gene expression profile of BL is similar to that of cells of the GC dark zone, whereas the expression profile of germinal center B-cell-like and activated B-cell-like DLBCL is more similar to that of light zone cells, suggesting that these lymphomas originate in different GC compartments.30 The relationship of BL to dark zone cells seems paradoxical because these cells do not normally express MYC.
Recent genomic sequencing studies have identified novel recurrent somatic mutations in BL.31,41,42 The most remarkable findings are frequent activating mutations in TCF3 and inactivating mutations in its inhibitor, ID3. The inactivation of ID3 likely impedes TCF3 in its inhibitory effect, resulting in a constitutive activation of this pathway (Figure 2A). ID3 mutations (38%-68%) are more frequent than those of TCF3 (11%). They are detected in ∼ 70% of sporadic and HIV-associated BL, but in only 40% of endemic tumors. These mutations are essential for the survival of BL cells and therefore constitute a necessary cooperating mechanism of MYC in the pathogenesis of BL.31 TCF3 and ID3 are expressed in the GC cells of the dark zone, but not the light zone, suggesting that mutations of these genes may retain the tumor cells in their differentiation compartment of the GC. The activation of TFC3 promotes the survival of BL cells (thus intensifying BCR signaling through the PI3K pathway) and their proliferation by up-regulating the expression of CCND3 (Figure 2A).31 Reinforcing the relevance of this mechanism, activating mutations of CCND3 have been detected in 38% of sporadic BL and occasional endemic tumors.31,41 In a recent mouse model, PI3K activation cooperating with MYC induces a lymphoma that resembles human BL, including the acquisition of CCND3 mutations.4 ID3 and TCF3 mutations have not been identified in DLBCL, reinforcing the idea that they are a cooperating mechanism of MYC to develop and maintain the identity of BL.
DLBCL with MYC translocations
Approximately 5% to 14% of DLBCL cases have been reported to carry MYC translocations.43,44 MYC amplification, although not systematically studied, has been reported in 2% of DLBCL cases in a recent study.45 An amplification of the translocated allele, a phenomenon named complicon, has been observed in some cases.46 Low copy number gains of MYC are more common in DLBCL (19%-38%) and may be associated with higher levels of miR expression.47 The presence of an underlying MYC translocation or amplification cannot be reliably predicted based on morphological and immunophenotypic features, although these cases tend to be positive for CD10 and BCL6 and may be negative for BCL2 (Figure 3A).45,48,49 In keeping with this, and in accordance with the concept of MYC translocations arising in the GC microenvironment, most MYC-rearranged DLBCL cases show a GC-type gene expression profile and/or a GC phenotype.40,49 MYC-rearranged DLBCL may either arise de novo or may represent a high-grade transformation of an antecedent low-grade lymphoma, most commonly follicular lymphoma (FL). In the latter case, the MYC gene rearrangement is frequently accompanied by a t(14;18)(q32;q21) chromosome translocation/BCL2 rearrangement. In larger series, ∼ 40% of patients whose tumors carried a dual MYC/BCL2 translocation were reported to have a history of, or were diagnosed with, concurrent FL.50 Conversely, 60% to 80% of MYC rearrangements in supposedly “bona fide” de novo DLBCL are accompanied by either BCL2 or BCL6 rearrangements, thus indicating that this “double-hit” (DH) scenario can also occur without clinical indication of a preceding low-grade disease (Figure 2A).44,49,51

Aggressive lymphomas with MYC translocations. (A) MYC-rearranged DLBCL. Note large blastic cells with broad cytoplasm and large nuclei, finely dispersed nuclear chromatin, and single prominent nucleoli (H&E stain; magnification, 400×). (B) BCLU. In comparison with BL, the tumor cells are slightly larger and harbor more irregular nuclei, sometimes with single nucleoli. Some histiocytes are interspersed, but there is no clearcut starry sky pattern (H&E stain; magnification, 400×). (C) PBL MYC rearranged. The cells are small to intermediate in size. Giemsa stain highlights the plasmablastic features of the tumor cells. CD20 was negative in this tumor (magnification, 1000×).
Aggressive lymphomas with MYC translocations. (A) MYC-rearranged DLBCL. Note large blastic cells with broad cytoplasm and large nuclei, finely dispersed nuclear chromatin, and single prominent nucleoli (H&E stain; magnification, 400×). (B) BCLU. In comparison with BL, the tumor cells are slightly larger and harbor more irregular nuclei, sometimes with single nucleoli. Some histiocytes are interspersed, but there is no clearcut starry sky pattern (H&E stain; magnification, 400×). (C) PBL MYC rearranged. The cells are small to intermediate in size. Giemsa stain highlights the plasmablastic features of the tumor cells. CD20 was negative in this tumor (magnification, 1000×).
In contrast to BL, MYC rearrangements in DLBCL are usually seen in the context of complex karyotypic alterations,40,52 and MYC is more frequently translocated to IGL or to non-IG genes such as BCL6, BCL11A, PAX5, or ICAROS49.53 MYC rearrangements in DLBCL have predominantly been demonstrated in patients > 60 years of age with higher clinical stage, higher International Prognostic Index values, and frequent presentation in extranodal sites, but these features are not consistent in all studies.44,51,54
A MYC rearrangement predicted an inferior outcome in DLBCL in most studies, but it is not yet entirely clear if this is due to the MYC rearrangement itself or because 58% to 83% of MYC-translocated DLBCL cases harbor dual or even triple translocations also targeting BCL2 and/or BCL6.45,49,51 Amplifications, but not low copy number alterations, have also been associated with shorter overall survival.45
MYC protein expression is seen in the majority of DLBCL cases, but the number of positive cells strongly varies from case to case.45,55 MYC protein is highly expressed (> 70% of cells) in the nuclei of DLBCL with MYC rearrangements or amplification. However, only one-third of DLBCL cases with substantial (> 30%-40% cells) MYC protein expression do carry MYC gene alterations.33,45,49,55,56 This suggests that mechanisms other than gene rearrangements are responsible for elevated protein expression in a considerable proportion of DLBCL cases.57,58 Protein overexpression of MYC has been associated with inferior prognosis in some studies,45 but as is the case with MYC translocations, MYC overexpression in DLBCL may not be predictive of an inferior prognosis on its own, because there is good evidence that it is the dual deregulation of both MYC and BCL2 expression that is strongly correlated with shorter survival.33,45,56,59 Immunohistochemical expression scores using MYC, BCL2, and possibly BCL6 are able to identify patients with poor prognosis even within International Prognostic Index subgroups.45,49,55,56
B cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL (BCLU) is a provisional category in the World Health Organization (WHO) 2008 lymphoma classification.60 BCLU is primarily defined by morphological features (Figure 3B). Interestingly, MYC rearrangements can be detected in 30% to 50% of these tumors.40,48,61 As the name implies, BCLU represents an “intermediate” category, with the tumors displaying features of both DLBCL and BL. The term “intermediate” lymphomas was derived from gene expression profiling studies in which tumors were classified as “molecularly intermediate” if their gene expression profile was fully consistent with neither BL nor DLBCL.40,62 BCLU is a disease of older patients presenting with nodal or extranodal disease usually in an advanced clinical stage.60 Both by morphology and by immunohistochemistry, some cases in this category bear more resemblance to BL, whereas others are more reminiscent of DLBCL (Table 3).
Morphological, immunological, and genetic features differentiating BL, BCLU, and DLBCL with MYC rearrangements

In contrast to BL, BCLU generally harbors many more aberrations in addition to 8q24 (MYC) translocations.50,63-65 The molecular structure of MYC translocations varies between BL and BCLU. In the former, MYC-IGH translocations prevail, whereas MYC is more frequently translocated to IGL or to non-IG genes in the latter.52 Although the exact frequency is not known, the MYC rearrangement is accompanied by translocations involving BCL2, BCL6, or both in ∼20%. These particular tumors are referred to as DH or “triple-hit” (TH) lymphomas. Gene expression profiling analyses have shown a profile intermediate between BL and DLBCL in some of the DH/TH lymphomas, whereas others display a BL gene expression profile.40,62 DH/TH lymphomas usually are aggressive neoplasms presenting with high clinical stage, high lactate dehydrogenase, frequent extranodal manifestations, and BM and CNS infiltration. A satisfactory therapeutic approach is lacking and the average survival time is short, usually < 1 year.50,63-65 Although the outcome of the patients in most, if not all, reports has been described as poor, cases with an antecedent or simultaneously occurring FL may fare still worse.65
DH/TH lymphoma
The DH/TH genetic constitution is not restricted to DLBCL and BCLU. It has also been observed in FL,66 and in B-cell lymphoblastic leukemia/lymphoma (TdT+).65,67,68 Most DH lymphomas harbor concomitant MYC and BCL2 gene translocations, but a minority of them have MYC/BCL6 rearrangements. In Mitelman's database of cytogenetic alterations in cancer, 62% of DH/TH cases harbor MYC/BCL2 translocations; a TH constellation involving MYC, BCL2, and BCL6 is encountered in 16%; and MYC/BCL6-rearranged cases account for only 8%.69,70 More recently, MYC/BCL6-rearranged lymphomas have been reported to be more often CD10-negative but IRF4/MUM1-positive, and cytogenetically less complex than their MYC/BCL2 counterparts.71 In general, in most studies, patients with DH/TH have been reported to run a dismal clinical course.50,54,63,64
Plasmablastic lymphoma and PCM
Plasmablastic lymphoma (PBL) is an aggressive neoplasm composed of a diffuse proliferation of large B cells, usually with immunoblastic morphology and the phenotype of a terminally differentiated B cell characterized by the loss of mature B-cell markers and expression of plasma cell–related antigens (Figure 3C).60 EBV infection with latency I is common but is not found in all cases. These tumors usually present in extranodal sites and frequently in the mucosae of the head and neck region in patients with different immunodeficiency states, mainly HIV infection.72-74 MYC translocations are encountered in 41% to 49% of PBL cases, virtually all of them with an IG gene as a partner and usually in the context of multiple chromosomal aberrations, and seem to confer an inferior prognosis.73-76
MYC activation also seems to play a role in the progression of plasma cell neoplasms, particularly from monoclonal gammopathy of undetermined significance to PCM. This progression is associated with increased levels of MYC expression in the absence of structural alterations of the gene.77 MYC rearrangements have been found in 0% to 15% of unselected PCM, but in 45% of advanced tumors, particularly in those with extramedullary involvement, and in 65% of PCM cell lines, suggesting that MYC structural alterations are associated with progression of the tumors.73,78,79 Contrary to PBL, MYC in PCM is frequently rearranged to non-IGH loci.79 The functional relevance of MYC in PCM has been highlighted by the addiction of these cells to MYC for survival.80,81
Some patients with PBL and MYC rearrangements have overlapping features with PCM,82 but the clinical context, immunodeficiency status, and EBV infection will help to distinguish these 2 entities. Secondary PBL transformed from chronic lymphocytic leukemia or FL also frequently harbor MYC translocations.83,84 All of these observations suggest that MYC aberrations do also play a role in the pathogenesis of aggressive lymphoid neoplasms with terminal B-cell differentiation. This finding is remarkable because most aggressive B-cell lymphomas with MYC rearrangements have a GC phenotype.
The terminal B-cell differentiation program is triggered by BLIMP1, a transcription factor highly expressed in PBL.85,86 BLIMP1 represses genes that maintain the mature B-cell identity, such as PAX5, and promotes the expression of genes involved in plasma cell differentiation, such as XBP1. BLIMP1 also represses MYC and other genes controlling cell proliferation and cell growth. The frequent presence of MYC translocations in these tumors may be required to overcome the repressing effect of BLIMP1 on MYC (Figure 2B). PCM, and probably also related neoplasias, have an active unfolded protein response, a protective antiapoptotic mechanism triggered in the endoplasmic reticulum that ensures the proper handling of the high protein load produced in these cells.87 This protective mechanism may help to bypass the proapoptotic effect of MYC. Interestingly, MYC oncogenic activation also seems to promote the unfolded protein response in transformed cells as a mechanism to escape from its apoptotic effects.88
Anaplastic lymphoma kinase–positive large B-cell lymphoma
Anaplastic lymphoma kinase (ALK)–positive large B-cell lymphoma is an aggressive tumor composed of immunoblasts with a plasmablastic phenotype and expression of ALK protein due to activating gene rearrangements with different partner chromosomes.89 These tumors lack mature B-cell markers and express BLIMP1 and XBP1. Contrary to other PBL types, these tumors do not carry MYC translocations, but express high levels of MYC protein.90 The mechanism activating MYC in these tumors is not clear, but may be a consequence of STAT3 activation. STAT3 is a downstream effector of ALK and is phosphorylated in ALK-positive LBCL.73,91,92 STAT3 also induces the expression of BLIMP1, promoting plasma cell differentiation (Figure 2B). Similar to PBL, the activation of MYC by STAT3 may be a mechanism to overcome the repressing effects of BLIMP1.
Summary and perspectives
The recent elucidation of the function of MYC in the development of the GC has provided a framework for us to also better understand its role in lymphomagenesis. The availability of new FISH probes and antibodies have facilitated the study of MYC alterations in aggressive lymphomas. MYC translocations are a diagnostic feature of BL and, in this disease, are frequently associated with a simple karyotype and somatic mutations activating the TCF3/ID3 pathway. In contrast, MYC gene alterations seem to represent secondary events associated with complex karyotypes in large B-cell lymphomas and are frequently associated with BCL2 or BCL6 translocations that confer a remarkable aggressiveness to the tumors. Several large B-cell lymphomas have MYC protein up-regulation independent of gene alterations. The concomitant overexpression of BCL2 protein in these tumors is associated with poor prognosis. Although most studies concur on the prognostic value of these “double” genetic or immunohistochemical “hits,” it is not completely clear if both have a similar significance. Further studies are needed to clarify how these new findings should be incorporated in the clinical setting. Immunohistochemical studies are easier to perform than genetic analyses. However, the difficulties in reproducing quantitative scores for some markers93 may preclude their routine application, suggesting that a screening approach using immunohistochemistry combined with FISH studies may be a helpful strategy.49 Although BL and DLBCL are distinctive lymphoma entities, molecular and pathological studies have recognized a subgroup of very aggressive tumors with intermediate features that are difficult to classify in these well-defined categories. These intermediate lymphomas (BCLUs) are a diagnostic challenge and their clinical and biological significance is not completely clear. All of these tumors are difficult to control with current therapeutic strategies. The integration of the new genetic and molecular diagnostic tools with novel treatment regimens and drugs may help to overcome the dismal prognosis of these malignancies.
This article was selected by the Blood and Hematology 2013 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2013. This article is reprinted with permission from Blood. 2013; Volume 122.
Acknowledgments
This work is supported by the Spanish Ministry of Science and Innovation (SAF2008-03630 and SAF2012-38432), Red Temática de Investigación Cooperativa del Cáncer (RD06/0020/0039 and RD12/0036/0036), Generalitat de Catalunya (2009-SGR-992 to E.C.), and the Robert Bosch Stiftung, Stuttgart, Germany (to G.O.). E.C. is an an Academia Researcher of the Institució Catalana de Recerca i Estudis Avançats of the Generalitat de Catalunya.
Disclosures
Conflict-of-interest disclosure: G.O. and A.R. declare no competing financial interests. E.C. has received honoraria from Pfizer and Janssen. Off-label drug use: None disclosed.
Correspondence
Elias Campo, Hematopathology Section, Department of Anatomic Pathology, Hospital Clinic, Villarroel 170; 08036-Barcelona, Spain; Phone: 34-93-2275450; Fax: 34-93-2275572; e-mail: ecampo@clinic.ub.es.
