
Abstract
Our understanding of the genetic basis of childhood acute lymphoblastic leukemia (ALL) has been greatly advanced by genomic profiling and sequencing studies. These efforts have characterized the genetic basis of recently described and poorly understood subtypes of ALL, including early T-cell precursor ALL, Philadelphia chromosome–like (Ph-like) ALL, and ALL with intrachromosomal amplification of chromosome 21, and have identified several rational therapeutic targets in high-risk ALL, notably ABL1-class and JAK-STAT inhibitors in Ph–like ALL. Deep sequencing studies are also refining our understanding of the genetic basis of clonal heterogeneity and relapse. These studies have elucidated the nature of clonal evolution during disease progression and identified genetic changes that confer resistance to specific therapeutic agents, including CREBBP and NT5C2. Genomic profiling has also identified common and rare inherited genetic variants that influence the risk of developing leukemia. These efforts are now being extended to ALL in adolescents and adults with the goal of fully defining the genetic landscape of ALL to further improve treatment outcomes in high-risk populations.
Learning Objectives
To understand how molecular approaches, including genome sequencing, have refined the classification of ALL
To appreciate variation in prevalence of ALL subtypes with age
To understand that specific genetic alterations are associated with treatment failure and are potential targets for therapy
To appreciate the relationship between clonal diversity and relapse
To understand the role of inherited genetic variation and risk of developing ALL
Childhood acute lymphoblastic leukemia
Acute lymphoblastic leukemia (ALL) is the most common pediatric tumor and, despite event-free survival rates now exceeding 85%, remains the leading cause of cancer-related death in children and young adults due to the often intractable nature of ALL relapse.1 Although the prevalence of ALL declines with increasing age, the outcome of treatment of ALL in adults is inferior to that of childhood ALL.2 This is only partly explained by the reduced frequency of genetic alterations associated with favorable outcome in children such as high hyperdiploidy and ETV6-RUNX1 and a rising incidence of adverse genetic alterations such as BCR-ABL1. Furthermore, there are remarkably few therapies targeted to specific genes or pathways and these are urgently needed in view of the dose-limiting toxicities of existing combination chemotherapy.
Over the last decade, there have been extensive efforts to use genome-wide profiling of genomic alterations in ALL to: (1) identify additional subtypes of ALL in cases lacking known aneuploidy or chromosomal rearrangements on cytogenetic analysis; (2) characterize the constellations of genetic alterations that define each ALL subtype; (3) identify the nature of clonal heterogeneity and how it influences treatment resistance and relapse; (4) define the role of inherited genetic variants in ALL susceptibility; and (5) translate these findings to improved diagnostic, prognostic, and targeted treatment approaches. These studies initially used microarray profiling of structural genetic changes (using single nucleotide polymorphism arrays or array-based comparative genomic hybridization) and gene expression and Sanger sequencing of limited numbers of genes. Current studies are using next-generation sequencing, including exome sequencing, transcriptome sequencing, and whole genome sequencing, to define the landscape of genetic alterations in ALL comprehensively.3-9 This chapter reviews the current understanding of cytogenetic and molecular classification of ALL, with an emphasis on recent discoveries and insights in new entities of ALL, particularly those that may influence clinical practice.
Genetic subtypes of ALL
Recurring gross chromosomal changes are a hallmark of ALL, with variation in frequency according to age. In childhood B-progenitor ALL, these include high hyperdiploidy with >50 chromosomes; hypodiploidy with <44 chromosomes; and chromosomal rearrangements including t(12;21)(p13;q22) ETV6-RUNX1 (TEL-AML1), t(1;19)(q23;p13) TCF3-PBX1 (E2A-PBX1), and t(9;22)(q34;q11) BCR-ABL1, rearrangement of CRLF2 at the pseudoautosomal region 1 at Xq21.3/Yp11.2, and rearrangements of MLL. Ph-like ALL is a recently described subtype characterized by a gene expression profile similar to BCR-ABL1 [Philadelphia chromosome (Ph)–positive] ALL and a diverse range of kinase-activating rearrangements and mutations (see “Ph-like ALL”). T-lineage ALL is characterized by activating mutations of NOTCH1 and rearrangements of transcription factors TLX1 (HOX11), TLX3 (HOX11L2), LYL1, TAL1, and MLL.10
The prevalence of these subtypes varies significantly with age (Table 1). High hyperdiploidy and ETV6-RUNX1 ALL are far less common in adults than in children and the prevalence of BCR-ABL1 and Ph-like ALL rise with high-risk features and increasing age. It is important to note that much remains to be learned regarding the genetic subtypes of ALL. Even in childhood ALL, which has been the most extensively sequenced, at least 11% of cases lack a known, leukemia-initiating chromosomal rearrangement. This proportion rises in both T- and B-cell ALL with increasing age, and comprehensive studies examining the genetic basis of ALL in older adults are lacking.
Frequency of ALL subtypes according to age
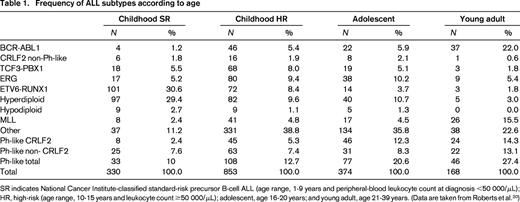
SR indicates National Cancer Institute-classified standard-risk precursor B-cell ALL (age range, 1-9 years and peripheral-blood leukocyte count at diagnosis <50 000/μL); HR, high-risk (age range, 10-15 years and leukocyte count ≥50 000/μL); adolescent, age 16-20 years; and young adult, age 21-39 years. (Data are taken from Roberts et al.20 )
Recurring alterations of multiple key pathways in ALL
Compared with many other tumor types in childhood and adults, childhood ALL has a relatively low mutation rate, with few nonsilent mutations and structural alterations per case. With the exception of the stereotyped aneuploidy characteristic of high hyperdiploid and hypodiploid ALL, chromosomal instability is uncommon. DNA copy number alterations are most commonly focal deletions and often arise from the activity of the antigen receptor gene recombinatorial machinery.8,11 Most ALL genomes have <10-20 nonsilent mutations per case, with exceptions being occasional relapse cases with hypermutator phenotypes. Indeed, MLL-rearranged infant leukemia has one of the lowest mutational frequencies of any described tumor.
Despite this low mutational load, key pathways are mutated at high frequency across multiple subtypes of ALL, including: (1) transcriptional regulation of lymphoid development (eg, PAX5, IKZF1, EBF1, ETV6, LMO2); (2) tumor suppression and cell cycle regulation (TP53, RB1, CDKN2A/CDKN2B); (3) cytokine receptor (CRLF2, EPOR, IL7R), kinase (ABL1, ABL2, CSF1R, JAK2, PDGFRB) and Ras (KRAS, NF1, NRAS, PTPN11) signaling; (4) lymphoid signaling (BTLA, CD200); and (5) epigenetic modification (EZH2, CREBBP, SETD2, MLL2, NSD2).12 Although alteration of these pathways is common across the spectrum of ALL, the specific genes involved and the type of alteration (eg, chromosomal rearrangement, deletion/amplification, or sequence mutation) vary substantially between subtypes. Moreover, specific genetic alterations are associated with the risk of treatment failure and relapse. For example, alterations of B-lymphoid transcription factor genes are a hallmark of B-ALL. PAX5 (paired box 5) alterations are frequent in B-ALL and are particularly common in subtypes such as ETV6-RUNX1 leukemia, but in most studies are not associated with treatment failure or relapse. In contrast, IKAROS gene family alterations are selectively associated with different subtypes of high-risk ALL. Expression of the founding member of this gene family, IKZF1 (IKAROS), is required for lymphoid development. Mutation of IKZF1 is observed in ∼15% of B-ALL cases and is a hallmark of BCR-ABL1 ALL and Ph-like ALL. IKZF1 alterations are independently associated with poor outcome in most studies of childhood and adult ALL, including BCR-ABL1 ALL, in the era of tyrosine kinase inhibitor (TKI) therapy.13 In contrast, alterations of IKZF2 (HELIOS) and IKZF3 (AIOLOS) are selectively associated with near haploid and low hypodiploid ALL, but are otherwise rare in non-hypodiploid ALL.4
Epigenetic alterations in ALL
Recent studies have implicated epigenetic deregulation in leukemogenesis and treatment failure. Cytosine methylation is an important mechanism regulating gene expression, with hypermethylation of CpG-rich regions in gene promoters associated with gene silencing and hypomethylation with transcriptional activation. Integrated genome-wide profiling of cytosine methylation, gene expression and deletions, and amplification of DNA have shown that established cytogenetic subtypes of ALL not only have distinct gene expression profiles, but also distinct methylation signatures and that methylation levels are significantly associated with expression in a substantial fraction of genes for each ALL subtype.14 Therefore, aberrant methylation is an important determinant of the leukemic transcriptome and is likely important in leukemogenesis.
Multiple recent studies have described mutations in genes that regulate the epigenome. These include genes that encode proteins that modify DNA or chromatin, erase or read modifications, or erase or read histones themselves. Important examples in ALL are genetic alterations of the polycomb repressor complex 2 (PRC2) that mediates repressive histone 3 lysine 27 (H3K27) trimethylation (most commonly EZH2, but also SUZ12 and EED) in early T-cell-precursor (ETP) ALL,3 WHSC1 (NSD2) alterations in ETV6-RUNX1 ALL,15 mutation of CREBBP (CREB-binding protein, a H3K18 and H3K27 acetylase) in relapsed16 and hypodiploid ALL,4 and mutations of SETD2 (a H3K36 trimethylase), KDM6A, and MLL2 in relapsed ALL.17 These findings are of clinical relevance because drugs that target histone readers (eg, bromodomain inhibitors) and histone modifiers (eg, histone demethylase inhibitors and histone deacetylase inhibitors) are in clinical trials and have been shown to be effective in experimental models of ALL.
Ph-like ALL
One of the most clinically important recent discoveries has been the characterization of Ph-like (also known as BCR-ABL1-like) ALL. This entity was identified by US18 and Dutch19 investigators using different gene expression profiling approaches. Ph-like ALL cases are BCR-ABL1 negative, but exhibit a gene expression profile similar to that of BCR-ABL1-positive ALL, have alteration of B-lymphoid transcription factor genes (most commonly IKZF1), and are associated with poor outcome. Ph-like ALL comprises 10%–15% of standard and high-risk childhood B-ALL, with an increasing prevalence with increasing age. Transcriptome sequencing (RNA-seq) of 15 cases identified rearrangements of kinase and cytokine receptor genes, including ABL1, EPOR, JAK2, and PDGFRB,5 suggesting that kinase-activating rearrangements are a hallmark of Ph-like ALL and that these alterations might be amenable to inhibition with approved TKIs.
In a recent study of >1700 childhood, adolescent, and young adult B-ALL cases, the frequency of Ph-like ALL was found to rise with age to 20.6% of B-ALL in adolescents and 27.4% of young adults (Table 1) and was associated with event-free and overall survival rates equal or inferior to high-risk ALL subtypes, including BCR-ABL1 positive and MLL-rearranged ALL.20 Fusion-specific assays and next-generation sequencing were used to characterize the genetic alterations activating kinase signaling in Ph-like ALL, which may be grouped into several categories (Figure 1, Table 2). The first category is ABL1-class rearrangements. These involve ABL1, ABL2, CSF1R (encoding the macrophage colony stimulating factor receptor), and PDGRB. Multiple fusion partners have been identified, but in each case, the fusion involves the kinase as the 3′ partner, preserving the kinase domain. The second category is JAK2/EPOR rearrangements. This group comprises JAK2 rearrangements with at least 10 fusion partners, also preserving the JAK2 kinase domain, and rearrangements of the eryothropoietin receptor with the immunoglobulin heavy (IGH) or kappa (IGK) loci that deregulate EPOR expression. The third category, CRLF2 rearrangements, are observed in ∼50% of Ph-like ALL and are often accompanied by JAK1 or JAK2 mutations and activation of JAK-STAT signaling. The fourth category is uncommon kinase alterations. A small number of Ph-like cases have rearrangements that have only been identified in single or few cases, including NTRK3 (also rearranged in other tumors), PTK2B, and the Janus kinase TYK2. The final category is Ras pathway mutations. A minority of Ph-like cases have no other genomic alterations activating signaling apart from those activating Ras signaling, including NRAS, KRAS, PTPN11, and NF1 mutations. Importantly, these mutations are not restricted to this subtype of Ph-like ALL and are also observed in other ALL subtypes, including high-hyperdiploid and MLL-rearranged ALL.

Genetic subtypes of Ph-like ALL. (A) Breakdown of Ph-like ALL into CRFL2-rearranged JAK mutant, CRFL2-rearranged JAK wild-type (WT), all other kinase lesions, and unknown. (B) Breakdown of “other kinase lesion” into the indicated subgroups based on genetic alteration. ABL1-class (ABL1, ABL2, CSF1R, PDGFRB); other JAK-STAT (FLT3, IL7R, SH2B3, JAK1/3, TYK2, IL2RB, TSLP); Ras (KRAS, NRAS, NF1, PTPN11, BRAF). HR indicates high-risk. (Adapted with permission from from Roberts et al.20 )
Genetic subtypes of Ph-like ALL. (A) Breakdown of Ph-like ALL into CRFL2-rearranged JAK mutant, CRFL2-rearranged JAK wild-type (WT), all other kinase lesions, and unknown. (B) Breakdown of “other kinase lesion” into the indicated subgroups based on genetic alteration. ABL1-class (ABL1, ABL2, CSF1R, PDGFRB); other JAK-STAT (FLT3, IL7R, SH2B3, JAK1/3, TYK2, IL2RB, TSLP); Ras (KRAS, NRAS, NF1, PTPN11, BRAF). HR indicates high-risk. (Adapted with permission from from Roberts et al.20 )
Kinase rearrangements and therapeutic targets in Ph-like ALL
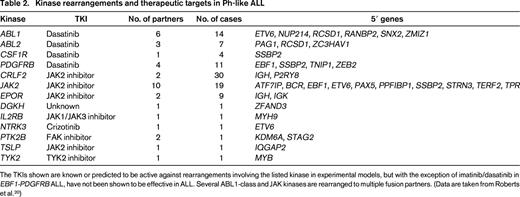
The TKIs shown are known or predicted to be active against rearrangements involving the listed kinase in experimental models, but with the exception of imatinib/dasatinib in EBF1-PDGFRB ALL, have not been shown to be effective in ALL. Several ABL1-class and JAK kinases are rearranged to multiple fusion partners. (Data are taken from Roberts et al.20 )
Data from cell lines expressing kinase fusions and xenografts and anecdotal case reports indicate that the majority of the Ph-like fusions induce activation of kinase-signaling pathways that is amenable to inhibition by available TKIs. Leukemic cells harboring ABL1, ABL2, CSF1R, or PDGFRB fusions are sensitive to ABL1 inhibitors such as imatinib and dasatinib at concentrations similar to BCR-ABL1-positive cells. Moreover, multiple patients have been identified with high-risk, relapsed, or refractory ALL harboring such fusions that have exhibited profound and durable responses to the addition of these agents.20,21 Because dasatinib has been safely and effectively combined with chemotherapy in the treatment of childhood BCR-ABL1 ALL, these findings have provided the rationale for prospective trials identifying children with Ph-like ALL and ABL1-class fusions and incorporating dasatinib therapy. There are also in vitro and preclinical xenograft data demonstrating the efficacy of Janus kinase inhibitors in Ph-like cases with JAK-STAT activation, particularly those with JAK2 rearrangements and IL7R/SH2B3 alterations, but clinical data demonstrating efficacy are not yet available.
Despite this genomic complexity, several approaches can be used to detect Ph-like ALL and the underlying kinase-activating lesions to inform diagnosis and treatment. It is likely that many centers will eventually use genome sequencing at diagnosis. Alternatively, screening and targeted approaches may be used, including low-density gene expression arrays to identify Ph-like cases, RT-PCR to detect known fusions, FISH for kinase disruption and specific fusions, and RNA or genome sequencing for cases lacking a known alteration.
CRLF2-rearranged ALL
CRLF2 rearrangements are common in Ph-like ALL, but are also observed in non-Ph-like ALL, particularly in >50% of Down syndrome ALL, in which IKZF1 mutations are less frequent.22 In both contexts, CRLF2 expression is deregulated by rearrangement to the IGH locus (IGH-CRLF2) or by a focal deletion 5′ of CRLF2 resulting in a P2RY8-CRLF2 fusion.22,23 CRLF2 rearrangement may be detected by FISH and CRLF2 overexpression by flow cytometry of leukemic cells. Approximately half of CRLF2-rearranged cases have concomitant activating sequence mutations of JAK1/2, most commonly at JAK2 p.Arg683 and many of the CRLF2-rearranged cases lacking Janus kinase mutations have other lesions that also activate JAK-STAT signaling, including IL7R mutations and deletions of the JAK-STAT-negative regulator SH2B3 (LNK). CRLF2-rearranged leukemic cells exhibit activated JAK-STAT, PI3K/mTOR, and BCL-2 signaling, and therapies targeting these pathways are being pursued in preclinical and clinical studies.
ERG alterations in ALL
Five to 10% of B-ALL cases exhibit a distinctive gene expression profile, but harbor few genomic alterations and no recurring chromosomal rearrangement. Approximately 75% of these cases have a focal deletion involving part of the ERG gene,24 which encodes an ETS-domain-containing transcription factor that is rearranged in prostate cancer and overexpressed in poor-risk acute myeloid leukemia. These cases often express an aberrant ERG protein. Of clinical importance, IKZF1 alterations are relatively common in ERG ALL, but, in contrast to BCR-ABL1 and Ph-like ALL, are not associated with inferior outcome.25 The genetic basis of cases in this subgroup that lack ERG deletions is unknown.
Intrachromosomal amplification of chromosome 21
Intrachromosomal amplification of chromosome 21 (iAMP21) occurs in up to 2% of childhood B-progenitor ALL, but is rare in adult ALL. In trials of childhood ALL in the United Kingdom, iAMP21 has been associated with older age and poor outcome.26 iAMP21 is defined by gain of at least 3 copies of large regions of chromosome 21, including RUNX1, and is most commonly observed as the sole chromosomal rearrangement. Less commonly, iAMP21 is observed in other ALL subtypes, including Ph-like and ETV6-RUNX1 ALL. Genome sequencing has demonstrated complex rearrangements that lead to sequential amplification and deregulation of genes in the amplified region.9
Hypodiploid ALL
Hypodiploidy with <45 chromosomes is associated with poor outcome and genomic analysis has characterized the 2 main subtypes of hypodiploid ALL: near haploid ALL, with 24-31 chromosomes, frequent Ras activating mutations, and alteration of IKZF3, and low hypodiploid ALL, with 32-39 chromosomes, alteration of TP53, CDKN2A/B, and/or RB1, and mutation of IKZF2.4 Importantly, half of childhood low hypodiploid ALL cases have inherited TP53 mutations, indicating that hypodiploid ALL may be a manifestation of Li-Fraumeni syndrome and should prompt clinical TP53 testing and counseling. Hypodiploid leukemic cells characteristically exhibit activation of PI3K/mTOR and MEK-ERK signaling that is sensitive to PI3K inhibitors (but not MEK inhibitors), suggesting that this may represent a therapeutic approach in this form of ALL.
T-lineage ALL
As in B-progenitor ALL, T-lineage ALL is characterized by a generally stable genome and subtypes defined by constellations of structural and sequence alterations that deregulate key cellular pathways. These include recurring translocations that deregulate transcription factors, often by rearrangement to T-cell antigen receptor loci. Sequence mutations and deletions involve genes that regulate T-cell development and tumor suppressor pathways, including NOTCH1, FBXW7, PTEN, RB1, and genes of poorly understood function, including PHF627 and WT1.
Recent studies have characterized ETP ALL, an immature neoplasm that lacks expression of several T-cell markers (CD1a, CD8, and weak/absent CD5) and exhibits aberrant expression of myeloid/stem cell markers.28 Many studies have shown that ETP ALL cases have poor outcome, although this is less striking in recent studies.29 Several groups described recurring mutations in 3 pathways in ETP ALL: hematopoietic development (RUNX1, IKZF1, ETV6, GATA3, EP300); Ras and cytokine receptor signaling (NRAS, IL7R, KRAS, JAK1/3, NF1, PTPN11, SH2B3), and chromatin-modifying genes, particularly PRC2 genes and SETD2.3,30-32 These findings have several biologic and clinical implications. The transcriptional profile of ETP ALL is most similar to normal hematopoietic rather than normal T-cell precursors, suggesting that ETP ALL may represent part of spectrum of primitive neoplasms that arise in progenitors that retain multilineage potential, including “near” ETP ALL (with normal CD5 expression) and possibly biphenotypic and bilineal ALL. The degree of similarity of the genetic basis of these entities to ETP ALL is currently unknown. The involvement of JAK-STAT and PRC2 pathways in ETP ALL suggests that JAK inhibition and/or chromatin-modifying agents may be therapeutically useful in ETP ALL and both have shown activity in preclinical models.
As in B-ALL, the full range of structural and sequence mutations in T-ALL remain to be defined, but ongoing efforts have identified new recurring targets of mutation, including mutations ribosomal proteins and CNOT3, which encodes part of a transcriptional regulatory complex.6 Transcriptome sequencing of T-ALL has identified novel mutations and chimeric fusions, including kinases that are also mutated or rearranged in B-ALL such as PTK2B (FAK) and JAK2.7 These results suggest that more detailed and comprehensive sequencing efforts are likely to identify new subtypes of T-ALL and therapeutic targets.
Genetic heterogeneity, clonal evolution, and relapse in ALL
It has been known for many years from cytogenetic analysis that the ALL genome is not static but evolves over time. The advent of microarrays and sequencing has allowed the nature of genetic heterogeneity in ALL and its relationship to clonal evolution and relapse to be characterized with precision. Analysis of matched leukemic cells at diagnosis and relapse has demonstrated acquisition of new deletions and mutations and loss of diagnosis-specific lesions at relapse, but with preservation of key alterations and commonality of antigen receptor rearrangements.32 In addition, relapse-acquired deletions and mutations can often be detected at low levels at diagnosis. These observations indicate that, in the majority of cases, the predominant diagnosis and relapse clones arise from a common “ancestral” or “preleukemic” clone that has acquired some of the genetic alterations required to establish leukemia, but then evolves down at least 2 lineages. Subsequent genome sequencing has delineated this clonal substructure and evolution and has made several additional observations.33 Many relapse-acquired lesions are enriched in specific pathways, including B-cell development (IKZF1), tumor suppression (TP53),34 Ras signaling, chromatin modification (CREBBP, SETD2),17 and drug metabolism (NT5C2).33,35 Several of these alterations are known to induce a more stem cell-like state (eg, IKZF1) or confer resistance directly to specific chemotherapy agents such as CREBBP and glucocorticoids16 and mutations in the 5′-nucleotidase gene NT5C2 and nucleoside analogs.35 Many cases commonly have multiple subclonal mutations involving the same pathway, gene, or residue at diagnosis, but initial therapy eradicates all but one clone that survives to propagate relapse. Relapse most commonly arises from a minor clone at diagnosis that subsequently acquires additional mutations that facilitate resistance to therapy. Finally, many mutations present in the predominant clone at relapse may be detected at early time points in therapy, which has implications for molecular monitoring, mutation identification, and the prediction of the risk of relapse. This is particularly relevant given the growing interest in the use of deep-sequencing approaches to monitor levels of minimal residual disease.36
Germline genetic variation and ALL risk
There has been historically limited evidence of a major role of inherited predisposition to developing ALL, with limited familial clustering and twin concordance explained by intrauterine transmission of leukemic clones. However, recent studies have implicated several common inherited variants and rare mutations in ALL susceptibility. Genome-wide association studies using microarrays to genotype millions of single nucleotide polymorphisms in ALL cases and ethnically matched controls have identified multiple susceptibility loci associated with ALL risk. The most reproducible associations have been in genes that are also targets of somatic genetic alteration in ALL: IKZF1, ARID5B, CEBPE, and CDKN2A.37,38 IKZF1 and CEBPE are transcriptional regulators and CDKN2A/B encodes the INK4/ARF family of tumor suppressors and cell cycle regulators, suggesting that the associated variants may influence gene expression and leukemogenesis. In addition, specific variants are associated with ALL risk and outcome in specific ethnic groups (ARID5B) and with specific subtypes of ALL (GATA3 and Ph-like ALL).39
Several reports have identified rare germline variations that affect gene structure and function directly in sporadic and familial ALL. Inherited TP53 mutations are a hallmark of low hypodiploid ALL. In addition, that study identified additional cases with germline mutations in Ras signaling and DNA repair that are likely to be pathogenic in ALL. Familial ALL kindreds are rare but can be exceptionally informative in identifying causal mutations. A recent study sequenced 2 unrelated kindreds with autosomal-dominant ALL. Remarkably, leukemic cells of all affected individuals harbored a novel germline PAX5 mutation, p.Gly183Ser, that was shown to attenuate the transcriptional activity of PAX5 incompletely.40 All patients also exhibited loss of the PAX5 wild-type gene by deletion of chromosome 9p in leukemic cells, indicating that transmission of this mutation is tolerable in the heterozygous state, but severe attenuation of PAX5 activity is required for leukemogenesis. This mutation was not detected in >30 additional ALL kindreds, so additional mutations are likely to contribute to leukemogenesis in familial ALL. Another notable example of direct associations between rare germline alterations and specific forms of leukemia is that of the markedly elevated risk of developing ALL with iAMP21 in individuals born with the rare constitutional Robertsonian translocation between chromosomes 15 and 21, rob(15;21)(q10;q10)c.9
Conclusions and future directions
The studies described herein illustrate the tremendous power of genomic analysis to identify the genetic basis of leukemogenesis and disease relapse in ALL. It is important to note that the number of comprehensively characterized whole ALL genomes is relatively small and ongoing discovery efforts sequencing large numbers of tumors of each subtype are required to fully define the inherited and somatic genomic landscape of ALL, to characterize clonal substructure, and to identify the full repertoire of alterations that contribute to treatment failure and relapse. These efforts will not only enable the development of more faithful experimental and preclinical models, but will also provide valuable data to inform results of clinical sequencing approaches. These data are likely to transform the nature of clinical diagnostic efforts, which must implement either targeted approaches informed by genomic studies or next-generation sequencing at diagnosis to accurately classify, risk stratify, and direct patients to logical targeted therapy.
Acknowledgments
This work was supported by the American Lebanese Syrian Associated Charities of St Jude Children's Research Hospital, the National Cancer Institute of the National Institutes of Health, the Pew Charitable Trusts, the American Society of Hematology, the American Association for Cancer Research, Stand Up To Cancer, and the St. Baldrick's Foundation. I thank members of my laboratory and colleagues at St Jude Children's Research Hospital, the Children's Oncology Group, and the National Cancer Institute TARGET consortium (https://ocg.cancer.gov/programs/target). I apologize to those whose work could not be cited due to space constraints.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Off label use of TKIs in ALL.
Correspondence
Correspondence: Charles G Mullighan, Department of Pathology, St Jude Children's Research Hospital, 262 Danny Thomas Place, Mail Stop 342, Room 4047C, Memphis, TN 38105; Phone: (901)495-5994; Fax: (901)595-5947; e-mail: charles.mullighan@stjude.org.
