
Abstract
Tyrosine kinase inhibitors, now numbering 5 for the treatment of Philadelphia chromosome–positive leukemia, have proven ability to reduce clonal disease burden rapidly, dramatically, and durably, especially in chronic myeloid leukemia in the chronic phase. Deep molecular remissions are likely in most chronic phase patients and expectations on timing of response have been developed, validated as best as possible, and evolved over time. Increasing attention has been given to the initial decline of Bcr-Abl1 transcripts and the ultimate depth of molecular remission, overshadowing but not displacing the traditional role of cytogenetic response. This chapter reviews the evolution of response milestones for chronic phase chronic myeloid leukemia and tries to answer the question of how little disease is too much.
Learning Objectives
To understand conventional and novel response milestones in CML
To differentiate benefits of cytogenetic and molecular response
To acknowledge points during treatment when therapy change may be considered
Introduction
The treatment of chronic myeloid leukemia (CML) has truly been revolutionized by the discovery, implementation, and expansion of the family of tyrosine kinase inhibitors (TKIs), beginning with the prototype Bcr-Abl1 inhibitor imatinib. Sentinel preclinical work1 proved the potential of selective inhibition of the kinase to abrogate the growth of tumors in mice and inhibit colony formation of Bcr-Abl1+, but not normal hematopoietic progenitors. With introduction of imatinib in clinical trials, initial dosing was expectedly based on phase 1 studies,2 in which limiting toxicity was not evident and the pragmatic goal was hematologic response. In short order, it became apparent that significant fractions of patients could achieve deep and lasting response with continued TKI therapy, particularly in the early chronic phase of CML.3 During the further development of imatinib and subsequent generations of Bcr-Abl1 kinase inhibitors for Philadelphia chromosome–positive (Ph+) leukemia and still ongoing today, optimization of dose to maximize response, desire to minimize both early and late toxicity, and focus on increasingly deeper thresholds of disease burden reduction have been the rule. In the last several years, the quest for finality of treatment has continued and an additional goal of “treatment-free remission” (TFR) has been defined as the next frontier.
Response thresholds and milestones during CML therapy
Initially in the evolution of Bcr-Abl1 kinase inhibitor therapy, response thresholds in CML were those historically defined in previous eras of nonspecific cytoreductive therapy, conventional cytotoxic chemotherapy and IFN-based therapy.4 These included hematologic response, in which blood count parameters reverted to normal or a nonelevated state and clinical findings (splenomegaly) resolved. Cytogenetic response (CyR) was the mainstay of correlative disease burden reduction, with majority reduction in detectable clonal Ph+ cells (MCyR) as a meaningful threshold and logically complete eradication of detection by karyotype [complete CyR (CCyR)] representing a major milestone5-7 and one that changed the natural history of CML for those who achieved it.
With imatinib clinical trials ongoing, it became apparent that response beyond that detectable by karyotyping was increasingly possible and further thresholds could be and would be crossed. Molecular response measurement of specific DNA sequences for the fusion protein Bcr-Abl1 by amplification of mRNA for the variably but predictably spliced fusion was measurable but not widely used in disease management in CML before the advent of TKIs.8 The benefits of molecular testing for CML include the ability to define further depth of response once CCyR occurs, as well as to measure disease burden from diagnosis to the deepest of remission. Major molecular response (MMR) was born from the International Randomized Study of Interferon and STI571, or IRIS trial,3,9 in which newly diagnosed patients with CML were randomized to treatment with imatinib or IFN-based therapy. Reduction in Bcr-Abl1 transcripts to 1/1000th that of untreated levels (≥3 log reduction) defined a new threshold below which events on study (loss of response and progression) were notably lower.9,10 With the advent of defining this threshold, increasing numbers of patients achieving high-quality remissions on TKI therapy for CML, and hematologic and CyR becoming more the norm than the exception, increasing focus was turned onto the depth and implications of molecular response in CML.
In the last 10+ years since MMR was deemed and proven to be a meaningful molecular threshold for CML patients on TKI therapy, the long-term performance of imatinib and shorter-term performance of newer agents have meant increasing depth of response beyond MMR for the majority of patients and a need to define further response landmarks beyond MMR. The functional capacity of quantitative PCR as it is used broadly (good-quality academic and commercial laboratories) is limited to measurement down to response levels of 4-5 logs below standardized untreated levels; reliable quantitation below this range is beyond the reach of standard technique. The use of 2-step nested PCR11 can allow for better qualitative assessment, and newer technologies such as digital PCR12 could offer reliable quantitation of smaller amounts of residual transcripts. In the current era of large numbers of patients with response beyond MMR and significant research into the potential for treatment cessation and TFR, the response threshold of complete molecular remission (CMR) was born13,14 and continues to find its place. CMR defines a response threshold at which no detectable transcripts are noted with a sensitivity of 4.5 logs below International Scale (IS) baseline. In an effort to redirect away from the potential misunderstanding of determining a response “complete” (which may imply absence or disease or cure), the MRD level of 4.5 logs below standard baseline on the IS scale, or 0.0032% (MR4.5), has been more widely used than the ‘CMR’ term.15
CCyR versus MMR: which matters more?
Threshold CyR has been a milestone for patients with CML before and throughout the TKI era. First demonstrated during trials of IFN versus the oral alkylators (hydroxyurea and busulfan)4 and IFN in combination with cytarabine versus IFN alone,5 achievement of major CyR (MCyR) prolonged the survival of patients with CML. Because early IFN-treated patients were followed longer, limited numbers of patients with stable CCyR drove efforts to better clarify levels and implications of residual disease. Although ultimately CCyR was itself associated with improved outcome after IFN-based therapy,5-7 it is perhaps the fact that pre-TKI therapies were unlikely to engender CCyR that led to the focus on MCyR and its prognostic significance as TKI therapy emerged and began clinical trials.
In the TKI era, MCyR remains a significant milestone, but is perhaps considered “in transit” to CCyR. CCyR has been deemed and stands firmly as a response milestone affording critical protection against transforming disease. During the IRIS trial, early landmark analyses of imatinib-treated patients confirmed a shorter survival free of progression to accelerated or blastic phase CML for patients not achieving a CCyR both at 12 and 18 months of therapy.3 Subsequent later analyses confirmed the protection against disease progression offered by CCyR and that risk of competing events rose for those achieving CCyR later in treatment.15 CyR was noted to occur proportional to Sokal risk score, with higher Sokal risk equating to lower odds of CCyR; however, once achieved, higher Sokal risk did not carry forward and lead to persistent risk once CCyR occurred, as was the case with IFN-based therapy.
The IRIS trial incorporated molecular monitoring in addition to cytogenetic monitoring. Further reanalysis of this landmark dataset, focusing on response in terms of quantitative PCR only correlated early and late molecular response during imatinib therapy with outcome.10,16 Early transcript reduction (<10% by 6 months) and subsequent reduction often noted to be likely equivalent to CCyR (<1% by 12 months) were noted to be associated with superior event-free survival and the lowest rates of progression to advanced phase disease. This reanalysis highlighted the controversy regarding the timing and added value of MMR, noting that those with ≤0.1% by 18 months (rather than 12 months, as proposed in earlier publications and guidelines) benefitted with no progression to advanced phase disease and improved event-free survival. Loss of CyR was noted to be more likely (26% vs 3%) for those without the added margin of MMR at 18 months. This dataset and its long-term findings shifted early molecular response into focus as a timely checkpoint and later molecular response into a slight blur as many emphasized the protection against progression of CCyR as the “cake” and the added benefit of MMR as only the “icing.”
The 3-month question (or perhaps the 3- to 6-month question)
Certainly, with the majority of patients on imatinib achieving threshold CyRs within the first year of TKI therapy, logic dictated closer scrutiny to speed the identification of the minority of patients not expected to do as well. As mentioned above, IRIS trial data analyzed molecular response and highlighted the benefit of “early molecular response.” Years earlier, the concept of “early molecular response” (EMR) had been touted in smaller patient series17 and emerging data concluded that earlier molecular response (3 months or even as early as 4 weeks) may help to discriminate both patients with high likelihood of success (such as subsequent MMR) and, more importantly, those destined to do more poorly. The concept of threshold reduction to 1-log or 2-log reduction within the first 3-6 months of therapy emerged at a time when correlation of molecular and CyR and the ability to interchange was evolving.
Over time, with the importance of molecular testing in the minimal residual disease state, a series of studies18-20 sought the optimal numerical threshold reduction to define EMR and opened the debate over its “weight,” timing (ie, 3 or 6 months' time allowance), and degree to which it would drive change in therapy. Multiple reports confirmed the 10% threshold and emphasized the prognostic value of this response by 3 months of therapy; additional analyses21-23 noted the merit of combined 3- and 6-month assessment to more precisely define treatment failure and to account for the potential to “catch up” between 3 and 6 months, which may preserve response and outcome expectations without change in therapy. As the role of a patient's initial transcript levels was reexamined, strategies to be more exacting and to “personalize” guidelines have emerged to account for variability at diagnosis and examine the fold-reduction and “halving time”23,24 of disease burden early in treatment to increase the predictive value and minimize premature therapy change. The potential shift forthcoming in view of early response and from what point it should be measured is highlighted, along with the schematized view of overall disease reduction and relevant milestones, in Figure 1. Widely accepted methods and further data validating individualized measure response kinetics are being sought to solidify the applicability and accessibility of this more granular approach to EMR.

‘Inverted iceberg’ schematic of CML burden and reduction over time. Arrows represent individual patient initial Bcr-Abl1 transcript level reduction. EMR=early molecular response; CHR=complete hematologic response; PCyR=partial cytogenetic response; CCyR=complete cytogenetic response; MMR=major molecular response; MR=molecular response (3,4,4.5,5–6 logs).
‘Inverted iceberg’ schematic of CML burden and reduction over time. Arrows represent individual patient initial Bcr-Abl1 transcript level reduction. EMR=early molecular response; CHR=complete hematologic response; PCyR=partial cytogenetic response; CCyR=complete cytogenetic response; MMR=major molecular response; MR=molecular response (3,4,4.5,5–6 logs).
Because most of the initial data were derived from imatinib-treated patients, subsequent trials of second-generation TKIs have added further data demonstrating that, irrespective of TKI, EMR should be a primary treatment goal and that it correlates with improved progression-free and overall survival.25-27 Most notably, the fraction of patients treated with second-generation TKIs who miss EMR milestone(s) is smaller; however, given the limitations on options to confidently and safely address these cases, guidelines allow for further time to confirm or deny treatment failure. What is missing at this time are crucial data to prove the value of intervention at 3 or 6 months; that is, bringing response back on track and which strategy to do so. Planned and ongoing trials have incorporated this pivotal question, intimately related to the crucial question regarding initial therapy: is imatinib with subsequent intervention to correct missed milestones no less effective than more potent TKI therapy at diagnosis to avoid missed milestones?
MR 4.5, the response formerly known as CMR, and its implications
In the eyes of patients, particularly early in the TKI era, addressing the question raised by the title of this chapter, how little disease is too much, is straightforward: the answer would be “any disease is too much.” Before trial data proving the potential for TFR and with functional cure only a glimmer of hope, patients early in the TKI era aimed for PCR negative, and in Canada even formed a “Zero Club” for those patients having an undetectable transcript level at least once (personal communication, Zavie Miller, Zavie's Zero Club, Ottowa, Canada, July 2001). As longer-term imatinib patients and increasing numbers of patients treated with nilotinib and dasatinib gained such depths of remission, the significance of CCyR and MMR were clarified and the mystique and draw of deep molecular remission morphed into response milestones. The current reality of stable deep remission for chronic phase CML treated with TKIs, in summary, appears to be more that of nonproliferative clonal disease reduced to levels not routinely measurable and predicted (and increasingly proven) to remain dormant for the foreseeable future.28 One of the most telling reports regarding evident yet nonproliferating disease came from the TWISTER study of the Adelaide group, in which patients who had ceased imatinib therapy after ideal remission and several years with undetectable Bcr-Abl1 transcripts all harbored persistence of the clone as measured by patient-specific and highly sensitive DNA PCR.29 So if not of eradication, what of deep molecular remission and its benefits?
Defining the importance of molecular response beyond MMR has been increasingly queried in several settings and remains a challenging yet important question. One clear dilemma is that such response thresholds lie at or below the limit of detection of the majority of assays used to assess patients' response, both in commercial and academic settings. The importance placed on deeper molecular response is based on the fact that such response is sought and deemed necessary for consideration for treatment interruption or cessation, be it for clinical trials of TFR, pregnancy, or other reasons, and that it has been postulated that achievement of CMR or MR4.5 may be associated with better long-term outcome than simply cytogenetic and MMR.
A study from the M.D. Anderson group of a large number of imatinib and second-generation TKI-treated chronic phase CML patients critically examined the impact of ‘undetectable’ molecular response (beyond MR4.5) among those with CCyR.30 Although overall a benefit was observed in transformation-free and overall survival, this was not sustained when adjusting for lead-time bias (greater time required to achieve undetectable status) by landmark analysis; in addition, the notion of ‘sustained MR4.5’ was not found to afford protection in this analysis. A second analysis by the Bordeaux/Lyon group examined the impact of CMR (MR4.5/undetectable transcripts confirmed over serial assessments ≥2 months apart) and did note improvement in event-free and failure-free survival for those with deeper molecular response compared with CCyR plus any other depth of molecular response (±MMR).31 Our own analysis (Portland group) noted, within a study of quantifying relevant transcript rise and predicting relapse, that the improvement from MMR to CMR (defined as MR4.0 and subsequent nested PCR undetectable) was associated with protection by improved relapse-free survival compared with MMR without CMR.32 The benefits of deeper molecular remission therefore remain controversial; the long-term follow-up of increasingly vast numbers of patients in deep remission should suffice to answer the question.
What might functional cure mean for CML?
CML therapy with TKIs represents an unprecedented paradigm for cancer treatment based on several principles: (1) initial therapy with oral targeted therapy; (2) maintenance of initial ‘induction’ or ‘primary’ therapy in the presence of response indefinitely (at least based on current algorithms); and (3) the forecast of ability to achieve a meaningful deep remission potentially equitable with a ‘functional cure’ in a significant minority to nearly half of patients. Principle #2, indefinite chemotherapy, has been and is currently the most ripe for disassembly and reexamination. Triggered initially by several case reports, it became evident that deep remission with subsequent treatment cessation may not lead to disease regrowth and several trials have examined this potential.33,34 The principle findings of initial studies have been remarkably similar: patients relapse rapidly (nearly exclusively in the first 6 months of treatment cessation) if it occurs; success of treatment cessation is ∼40–50%; and predictors of success with cessation appear to be lower Sokal risk at diagnosis and longer duration of treatment. What has begun to broaden is the scope of discontinuation trials, now encompassing patients with more notable levels of residual disease, such as those in the EUROSKI trial,35 in which MR4.0 is the required remission depth for consideration of discontinuation. Large efforts as well continue on behalf of pharmaceutical partners, particularly Novartis, in which an array of discontinuation trials are ongoing to query the benefits of primary nilotinib therapy or switch to nilotinib after imatinib with regard to TFR.
In addition, the definition of success after treatment cessation has migrated from the initial trials' threshold of molecular relapse (detectable disease generally in the 4- to 4.5-log reduction range) to the current concept that retreatment should occur with loss of MMR. Although, at face value, this may portend that “failure” of the experiment of cessation may carry with it a 1.5-log increase in clonal disease (MR4.5 to >MR3.0) and any risk incumbent therein, several studies have addressed the pragmatic reality of patients' MRD, its stability, and consideration for TFR trials. It may be that the stability of deep remission may have little to do with the success of treatment cessation; in the According to STIM (A-STIM) trial, patients who discontinued from a stable CMR and an ‘unstable CMR’ had equal chances of success.36 In a thoughtful analysis by the Hammersmith group, the challenge of defining a population suitable for TFR trials using stringent criteria while acknowledging the quantitative PCR assay specifics needed to confidently report repeated, sequential MRD levels below rock-bottom thresholds was revealed37 and, in their center, 25% or fewer of patients would meet such criteria. Therefore, the view of how deep and how stable one's remission need be before consideration of TFR has evolved. It embraces the somewhat unique feature of CML under TKI therapy, that someone may be “cured” while still with evidence of disease [a concept also proposed in patients with t(8:21) fusions identified in long term remission after AML38 ].
So how little is too much?
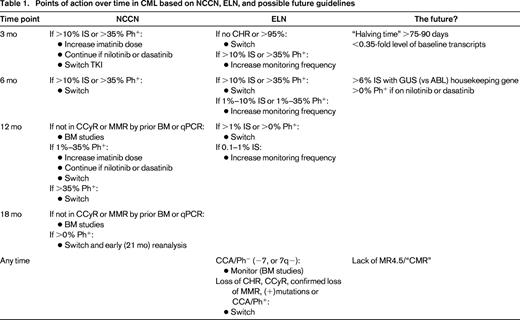
The lessons learned over the last 15+ years, during which time TKIs evolved from phenomenal yet uncertain to entrenched and unprecedented regarding their efficacy, have been that timely declines in Ph+ clonal burden below relevant thresholds anchored in history (CyR) and present research (kinetics of Bcr-Abl1 transcript decline over months 0-3) speak of “sensitive disease” and are required for safe passage into deep remission. Response is typically “biphasic” (rapid decline followed by slower decline) and in a sense gradual; of interest there does not appear to be a “ceiling effect” with regard to increasingly potent therapy used at initial diagnosis, as demonstrated by the preliminary data of ponatinib's activity in the now shuttered EPIC trial,39 with even higher rates of EMR and early MMR. Therefore, as long as early milestones are met, declining disease is acceptable. As noted above, caution is advised in calling response “failure” too early, such as in the case of seemingly inadequate early transcript reduction based on black and white thresholds (10% IS at 3 months), because individualization of response and accounting for initial disease burden may change perception and subsequent time point (eg, 6 months) improvement appears to mitigate initial risk of a missed or near-missed milestone. Nonetheless, guidelines are useful and necessary and both the National Comprehensive Cancer Network (NCCN) and European LeukemiaNet (ELN) algorithms are frequently updated and serve as a benchmark for best practice; from these, “action items” (Table 1) may be derived at which point disease burden, including very little at later time points, is viewed as too much and therapy change is warranted.
In essence, disease burden in CML is too much if it is permissive of clonal persistence, if it is permissive of instability and genesis of mutations, and of course if it is permissive of clonal expansion. Increasing understanding into the nature of stem cell biology and leukemogenesis, described by many as an accumulation of stem cell defects of varying potential impact characterized as either “passenger” or “driver” mutations, may turn the focus away from the quantity of residual disease but more on the quality of the remission. It may be that the benefit of better CML therapy is a result, not of eliminating more of it, but of eliminating the most unstable and proliferative elements and doing so expeditiously. It is unsettling to patients and dissatisfying to practitioners, but it appears that the “little” disease remaining in patients in deeper molecular remission may not be too much as stable responses are the expectation and treatment-free remissions appear to be a potential reality for many. Ongoing research to clarify the complex reality of TKI resistance and understand the behavior of residual CML after cessation of therapy, however, is needed before such truths become self-evident.
Disclosures
Conflict-of-interest disclosure: The author has consulted for Ariad, Pfizer, Bristol Myers Squibb, and Novartis Oncology. Off-label drug use: None disclosed.
Correspondence
Michael J. Mauro, MD, Memorial Sloan Kettering Cancer Center, 1275 York Ave., Box 489, New York, NY 10065; Phone: (212)639-3107; Fax: (212)772-8550; e-mail: maurom@mskcc.org.
