
Abstract
Over the past 4 decades, a better understanding of the genetic origins of inherited platelet disorders has illuminated avenues of investigation in megakaryopoiesis and has identified targets of pharmacologic intervention. Many of these discoveries have been translated into clinical medicine. The success of inherited platelet disorder research is underpinned by broader advances in methodology through the biochemical and molecular revolution of the 20th and 21st centuries, respectively. Recently, modern genomics techniques have affected platelet and platelet disorders research, allowing for the discovery of several genes involved in platelet production and function and for a deeper understanding of the RNA and miRNA networks that govern platelet function. In this short review, we focus on recent developments in the genetic elucidation of several disorders of platelet number and in the molecular architecture that determines the “genetic makeup” of a platelet in health and disease.
Learning Objective
To provide readers with recent research discoveries of the genetic causes of platelet disorders
Historical perspective
Nearly 100 years ago, Eduard Glanzmann, a Swiss pediatrician, first characterized a novel hereditary form of mucocutaneous bleeding,1 likely representing the first detailed scientific report of an inherited platelet disorder. In 1918, just 2 years into his own practice, Glanzmann coined the term “thrombasthenie” (weak platelets) to describe this malady with normal platelet count and morphology yet grossly abnormal clot retraction. This bleeding diathesis would become known as Glanzmann thrombasthenia (GT). Fifty years later, identification of multiple nuclear families and recently developed platelet aggregation techniques allowed further interrogation of thrombasthenic platelets, which demonstrated that these platelets were unable to bind fibrinogen and aggregate, even when physiologically stimulated.2,3
Over the next 2 decades, investigators further evaluated thrombasthenic platelets and determined that glycoproteins IIb (GPIIb) and GPIIIa were absent.4-7 The identification of GPIIb/IIIa led to development of monoclonal antibodies to further characterize this fibrinogen receptor.8-10 Subsequent monoclonal antibody design culminated in animal studies and successful clinical trials that prevented thrombosis and restenosis after percutaneous coronary intervention.11-14 Therefore, studies on families with GT expanded into the understanding of fibrinogen receptor structure and promoted the development of GPIIb/III inhibitors that revolutionized antithrombotic therapy. Based on this successful story, it is reasonable to think that the discovery of genetic and biological processes that are involved in congenital platelet disorders will lead to a better understanding of megakaryocyte and platelet biology and this will potentially translate into the design of better diagnostic tools and novel therapies.
Peptide and biochemical analysis not only helped elucidate the etiology of GT, but also the genetics of Bernard-Soulier syndrome, which is caused by mutations in GP9, GP1BA, or GP1BB, which encode components of the platelet GPIB-IX-V receptor that interact with the large multimeric glycoprotein VWF.15 Later characterization of DNA and the development of positional cloning and DNA amplification protocols provided the technology to identify several other genes now implicated in inherited platelet disorders, includingFLI1, GATA1, HSP1-HSP9, MYH9, RUNX1, and WAS.
Classification
Congenital platelet disorders are usually classified by platelet phenotypic parameters that often include morphology and size as well as mode of inheritance. This is particularly useful for the practicing hematologist when a diagnosis of a hereditary platelet disorder is being considered. More recently, with increased understanding of the mechanisms of megakaryopoiesis and platelet biology, inherited platelet disorders may also be classified by the megakaryocyte pathway affected by the mutated gene responsible for the disease (Figure 1). This molecular classification is complementary to the clinical classification and may allow scientists to not only expand their knowledge about pathophysiology, but also to ask fundamental questions about similar pathways affected in other cells and potential targeted therapies. Two recent articles provide a mechanistic review of inherited platelet disorders.16,17

Schematic figure of a megakaryocyte with pathways and potentially related genes that, when mutated, are associated with syndromes that include thrombocytopenia. Each box title represents a specific pathway or family of molecules with associated syndromes. Genes are italicized.
Schematic figure of a megakaryocyte with pathways and potentially related genes that, when mutated, are associated with syndromes that include thrombocytopenia. Each box title represents a specific pathway or family of molecules with associated syndromes. Genes are italicized.
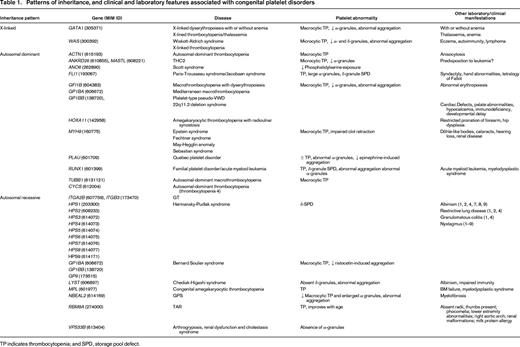
It is important to mention that several of these mutations also cause other clinical manifestations (Table 1), allowing clinicians to narrow the diagnosis of an inherited platelet disorder.
Recent developments
With the completion of the Human Genome Project in 2003 and vast advances in sequencing technology, genome-wide research became available in a more cost-effective manner and several Mendelian disorders began to be investigated in a nonbiased approach. Other massive collaborations, including the International HapMap project, the 1000 Genomes project, and the ENCODE project, provided increased understanding of the architecture, variation, and function of the human genome. These resources have allowed more accurate analysis of common variants and mapping of duplications and deletions to assess copy number variations.
In terms of platelet gene discovery, gray platelet syndrome (GPS), an autosomal-recessive α-granule storage disorder with macrocytic thrombocytopenia and myelofibrosis, is perhaps the best representation of the bridge between the biochemical and the genetics era. Although GPS had been phenotypically and biochemically fairly well characterized, the genetic cause for the disorder remained elusive for decades. Using homozygosity mapping, the GPS locus was initially mapped to chromosome 3p21.18,19 Simultaneous efforts using exome sequencing and RNA transcriptome sequencing identified biallelic mutations in NBEAL2 as the cause of GPS.20-22 NBEAL2 encodes a BEACH-domain containing protein with homology similar to the protein encoded by LYST.22 Recently 2 nbeal2−/− mouse models were shown to have defective α-granule biogenesis, resulting in impaired platelet adhesion, aggregation, and coagulant activity, as well as protection against thromboinflammatory brain infarction.23,24 NBEAL2, as well as VPS33B and VPS16B, which are also associated with α-granule deficiencies, are anticipated to participate in α-granule biogenesis, providing the first anchor points in the previously unsolved pathway.25
GFI1B, which encodes for the transcriptional repressor growth factor independent 1B was also recently implicated in a family with an autosomal-dominant form of GPS.26 Sequence analysis of GFI1B identified a familial nonsense mutation resulting in a truncated peptide. The truncation is located within a DNA-interacting zinc finger domain. It was further shown that the truncated peptide does not repress Gfi1b target expression in mice and has a dominant-negative effect on wild-type Gfi1b, inhibiting its normal function and therefore preventing normal megakaryocyte development and platelet production.
Thrombocytopenia with absent radius (TAR) syndrome is an example of a rare inherited platelet disorder in which the underlying gene was identified by exome sequencing.27 TAR was originally identified as a rare chromosome 1q21.1 microdeletion syndrome.28 However, unaffected parents also frequently showed the microdeletion, so it was clear that other genetic events had to be involved to trigger the phenotype. Exome sequencing in patients without the pathognomonic microdeletion demonstrated that complex heterozygosity in RBM8A, which encodes Y14, was sufficient to cause TAR. Therefore, most patients with TAR have a rare null allele at the RBM8A locus (usually involved in the microdeletion) and a low-frequency regulatory variant in the other allele. This complex genetic combination appears to be sufficient to cause TAR. Y14 is a RNA-binding protein that has been implicated in NF-κB-mediated transcription and apoptosis.29,30 Although mutations in RBM8A or the 1q21.1 microdeletion result in low levels of Y14, the mechanism underlying defective TPO signaling and thrombocytopenia is still unclear.
In addition to improved sequencing methodology, recent genetic advances have also provided invaluable analytic resources, including the 1000 Genomes project.31 Thrombocytopenia 2 (THC2), an autosomal-dominant form of thrombocytopenia, was also localized to chromosome 10p11.2-12 in 2 large unrelated Caucasian kindreds from Italy and the United States.32,33 Candidate gene sequencing proved inconclusive because mutations were reported in ACBD5 and in MASTL, respectively.34,35 Further screening of MASTL and ACBD5 mutations was negative in 4 Italian pedigrees, in which linkage to the THC2 locus was established.36 Successively, sequencing of 32 regional candidate genes identified 6 mutations in a conserved 19 bp region of the 5′-untranslated region (UTR) of ANKRD26 in 9 Italian families, including the original kindred with the putative ACBD5 mutation. Further sequence analysis identified a total of 12 candidate mutations confined to a 22 bp region of the 5′-UTR in 21 of 210 families with inherited thrombocytopenias of unknown cause.37 Comparison with the 1000 Genomes dataset confirmed polymorphisms in this 5′-UTR region that are not found in the general population. Interestingly, ANKRD26 5′-UTR mutations have also been associated with a predisposition for myeloid malignancies in conjunction with familial thrombocytopenia.38,39 The role of ANKRD26 in megakaryopoiesis remains unclear. However, it was proposed recently that THC2 might be caused by abnormal MAP kinase signaling, in which 5′-UTR mutations in ANKRD26 result in loss of RUNX1 and FLI1 protein binding, both transcription factors that were implicated previously in inherited platelet disorders.40
Quebec platelet disorder, another autosomal-dominant inherited platelet disorder with linkage to chromosome 10 (10q24), is characterized by a gain-of-function defect in fibrinolysis and increased platelet stores of urokinase plasminogen activator (uPA), which results in delayed-onset bleeding after clinical challenge.41-44 Although platelets are structurally normal and platelet counts minimally reduced to normal, there is plasmin-mediated degradation of α-granule proteins and increased uPA released from activated platelets in Quebec platelet disorder, resulting in decreased aggregation and accelerated clot lysis.45 Sequencing of the candidate gene PLAU, which encodes uPA, did not reveal putative coding or regulatory mutations, although it was observed that there was disproportionate allelic expression in patients, suggesting a cis regulatory defect.43 Copy number variation screening and subsequent targeted sequencing demonstrated a 78 kb tandem duplication of PLAU in patients, but not control samples.46 The mechanisms of mild thrombocytopenia and platelet dysfunction have not yet been elucidated. For example, it is unknown why up to 150-fold overexpression of PLAU in differentiating megakaryocytes with the tandem duplication does not translate into changes in plasma and urinary uPA in patients with this disease.
Although many inherited platelet disorders have been explained, the combination of traditional and modern biochemical and molecular techniques remains essential. This is the case in discovery of the gene that causes Scott syndrome, an autosomal-dominant bleeding diathesis in which platelet count, size, and aggregation are normal.47,48 When the platelets are activated, they fail to expose phosphatidylserine, a potent trigger of the coagulation cascade. Therefore, factor Va and Xa binding is decreased, resulting in decreased capacity to convert prothrombin to thrombin. Platelets in Scott syndrome were found to be deficient in calcium-dependent scrambling of phospholipids mediated by TMEM16F, a transmembrane calcium channel.49 Subsequent mRNA analysis in model cell lines allowed mapping of TMEM16F to the gene ANO6; sequencing of patient DNA identified splice site mutations resulting in premature termination of the protein.
Future of platelet genetic research
High-throughput genotyping platforms developed in the past decade have made it possible to screen for multiple mutations simultaneously. Efforts are ongoing to customize high-throughput screening assays for patients with mild bleeding disorders.50 Other studies have focused successfully on screening for candidate genes in families with history of excessive bleeding, including a recent report in which a enrichment of FLI1 and RUNX1 mutations were found in individuals with platelet secretion defects.51 Eventually, the mutational profile of patients with genetic thrombocytopenias and disorders of platelet function will be elucidated.
Perhaps one of the most fascinating recent discoveries in platelet research is that what was thought to be “vestigial” RNA in platelets actually appears to represent a complex network of mRNA and microRNA that may regulate thrombopoiesis and platelet function.52 Moreover, platelets have been shown to have an active spliceosome, indicating that mRNA processing occurs in platelets even without the presence of a nucleus.53 Therefore, the genetic makeup of a platelet may be determined at the platelet's “birth” and may explain the observed variability in platelet responses among healthy individuals. In addition, specific mRNA expression profiles have been associated with different diseases, such as cardiovascular disease and sickle cell anemia, raising the attractive hypothesis that megakaryocytes may respond to certain disease states by altering their transcriptome profile, delivering platelets with a specific molecular signature.54
Finally, this newly recognized platelet genetic diversity may have direct implications on platelet reactivity, predisposition to disease, and response to antiplatelet therapy. Recently, Edelstein et al55 showed that increased PAR4-induced platelet aggregation and calcium mobilization in blacks (compared with white healthy subjects) was associated with elevated phosphatidylcholine transfer protein (PCTP) mRNA levels and inversely correlated with levels of miR-376c, an miRNA that targets PCPT, the gene that encodes PCTP. This particular miRNA regulates expression of PCTP in human megakaryocytes, supporting a direct link between these RNA networks and platelet hyperreactivity in blacks.
In summary, the genetics of platelet disorders has evolved from the discovery of genes by traditional positional cloning to the use of modern genetic techniques such as whole exome/genome sequencing. This has not only expanded the repertoire of genes responsible for platelet disorders, but has also improved our understanding of platelet biology and megakaryopoiesis in general.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Jorge Di Paola, MD, University of Colorado School of Medicine, 12800 East 19th Ave., Aurora, CO 80045; Phone: (303)724-4000; Fax: (303)724-4015; e-mail: Jorge.dipaola@ucdenver.edu.
