
Abstract
Blood types (blood group antigens) are heritable polymorphic antigenic molecules on the surface of blood cells. These were amongst the first human Mendelian traits identified, and the genetic basis of nearly all of the hundreds of blood types is known. Clinical laboratory methods have proven useful to identify selected blood group gene variants, and use of genetic blood type information is becoming widespread. However, the breadth and complexity of clinically relevant blood group genetic variation poses challenges. With recent advances in next-generation sequencing technologies, a more comprehensive DNA sequence-based genetic blood typing approach is now feasible. This chapter introduces the practitioner to high-resolution genetic blood typing beginning with an overview of the genetics of blood group antigens, the clinical problem of allosensitization, current blood type testing methods, and then discussion of next-generation sequencing and its application to the problem of genetic blood typing.
Learning Objectives
To understand the different types of genetic variation
To introduce principles of the nomenclature used to describe DNA variants
To understand the strengths and limitations in assigning blood types using genetic information
Genetics of blood type
Blood group antigen (blood type) differences between donors and patients are the result of genetic variation in blood group genes that change the surface expression of blood cell antigens. To be assigned a number by the International Society for Blood Transfusion (ISBT) Working Party, a blood group antigen must be shown to be inherited.1 There are >300 different blood group antigens that are encoded by >40 blood group genes (select blood group systems are listed in Table 1). Blood group genes encode molecules spanning a wide range of structures and function (Figure 1), from carbohydrate moieties (ABO), to cytokine receptors (Duffy), to urea transporters (Kidd), to structural glycophorins (MNS).1 The blood group gene loci collectively exhibit all types of genetic variation, including single nucleotide variants (SNVs), insertions/deletions (indels), and structural variants (SVs; types of genetic variation are illustrated in Figure 2). Identification of blood group variants can be challenging. In any single individual, blood group variants generally result in subtle or no perceptible phenotype and thus are often of little clinical significance in the absence of an allogenic exposure. Furthermore, the frequencies of most blood group antigens vary between different ethnic populations, which can confound targeted assays. Curated and detailed DNA-phenotype annotated databases, such as those maintained by Database Red Blood Cells (dbRBC; Blood Group Antigen Gene Mutation Database), RhesusBase, National Center for Biotechnology Information, and ISBT, inform calling blood type from genetic data, but these databases appear to be biased with regards to ethnicity when compared with the blood group gene variants deposited in other databases2,3 and systematically underrepresent some types of blood group variation, particularly large insertions, deletions, copy number variation, and SVs. The two most clinically significant blood group systems, ABO and Rh, are both genetically complex and therefore provide useful examples of the significance of blood group gene variation.
Table of blood group systems, blood group antigens, frequencies, availability of antisera and SNP testing, and blood group genes and locations
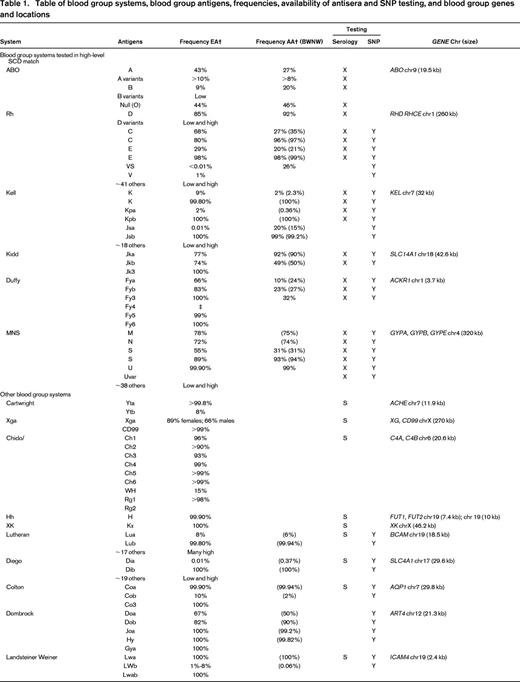
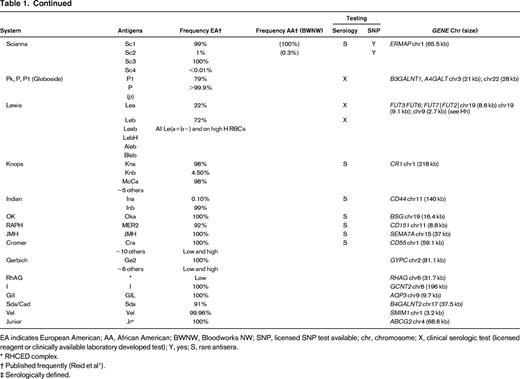
EA indicates European American; AA, African American; BWNW, Bloodworks NW; SNP, licensed SNP test available; chr, chromosome; X, clinical serologic test (licensed reagent or clinically available laboratory developed test); Y, yes; S, rare antisera.
* RHCED complex.
† Published frequently (Reid et al1 ).
‡ Serologically defined.

Illustration of blood group systems. There are >30 blood group systems that collectively include structurally diverse molecules that present blood group antigens on the surface of RBCs. Shown here are general schematics of the structures of major blood group systems that include carbohydrates, single-pass transmembrane proteins, multipass transmembrane proteins, and glycosylphosphatidylinositol-linked proteins.
Illustration of blood group systems. There are >30 blood group systems that collectively include structurally diverse molecules that present blood group antigens on the surface of RBCs. Shown here are general schematics of the structures of major blood group systems that include carbohydrates, single-pass transmembrane proteins, multipass transmembrane proteins, and glycosylphosphatidylinositol-linked proteins.

Types of DNA sequence variation. (Top) SNVs result from the substitution of one base, whereas insertion or deletion (indel) affects a string of nucleotides. (Bottom) SVs (typically affecting >1000 bp) include large indels, inversions, translocations, and CNVs. Reprinted with permission from Johnsen et al.39
Types of DNA sequence variation. (Top) SNVs result from the substitution of one base, whereas insertion or deletion (indel) affects a string of nucleotides. (Bottom) SVs (typically affecting >1000 bp) include large indels, inversions, translocations, and CNVs. Reprinted with permission from Johnsen et al.39
ABO is a carbohydrate blood group system encoded by a single glycosyltransferase gene, ABO (for review, see Storry and Olsson4 ). The ABO gene seems deceptively simple, spanning only 7 exons, and the majority of known ABO genetic variants are also seemingly relatively simple, primarily comprising SNVs and a handful of small indels. The genetic complexity of the ABO blood group system lies in the sheer number of these DNA variants, comprising >200 named ABO alleles. The challenge of assigning ABO blood type from this large number of ABO alleles is further confounded by the fact that the ABO gene does not encode ABO blood type antigens directly, but rather ABO encodes the ABO glycosyltransferase enzyme, which in turn makes ABO antigens. Thus, predicting the ABO blood group antigens from ABO genetic data requires deep knowledge of the spectrum of ABO alleles and understanding of the effect of variation on the function of the ABO glycosyltransferase. For ABO, the clinical significance of accurate assignment of ABO alleles cannot be overstated, because ABO mismatched transfusion can lead to life-threatening hemolytic transfusion reactions (HTRs).4 Serology-based ABO tests are inexpensive, highly sensitive, and generally sufficient to inform routine transfusion practices. However, ABO serologic discrepancies, when they occur, require additional testing. Additionally, in some clinical scenarios (platelet transfusion, transplant), knowledge of ABO subtype can guide clinical decision making.5 Predicting blood group O alleles is seemingly straightforward, because any DNA variant that disrupts ABO gene expression will result in an “O” allele.6 However, ABO variants can also change ABO glycosyltransferase specificity from the “A” (GalNAc-transferase) isoform to the “B” (galactose-transferase) isoform or vice versa or may simply direct a weakly functional ABO glycosyltransferase that can result in clinical ABO phenotype ambiguity. Expression of ABO can also rarely be ablated by loss of function of the precursor FUT1 (and FUT2) gene(s) independent of the ABO gene itself. Thus, in genetically assigning ABO, all 3 genes are considered. In the case of the ABO blood group system, there is ABO, FUT1, and FUT2 allele data in databases, such as dbRBC,7 which has alleles annotated with corresponding ABO serologic phenotype data for use in assigning genetic ABO blood group antigens.
The genetic diversity of the Rh blood group system lies in the structure of the RH genetic locus, which spans two paralogous neighboring genes, RHD and RHCE. There are >200 RHD alleles and >80 RHCE alleles that exhibit all types of genetic variation, including SNVs, indels, and, notably, SVs. The most common cause of D-negative blood type is a whole RHD gene deletion, although other gene disrupting mutations also occur. Alleles likely resulting from interlocus gene conversion between RHD and RHCE are also common, leading to RHD–RHCE hybrid alleles that can affect antigen presentation.8 Rh typing is often simplified to RHD (RhD) and D positive versus D negative. D-negative blood type has high prevalence (15%–17%) in European Americans and is also present in African ancestries (2%–5%) but is relatively rare in Asians (<0.1%).8 In patients at high risk for allosensitization, such as in multiply transfused sickle cell disease patients, additional consideration of the RHCE variants C, c, E, and e is recommended.9 However, the Rh blood group system is far more complex than D and CcEe, with >50 serologically defined antigens described, reflecting the underlying genetic diversity at the RH locus. To clinically categorize RHD phenotypic diversity, RHD variants encoding proteins that exhibit common D epitopes but are lacking others are termed “partial D,” whereas variants that result in intact but overall weak expression of D on the RBC surface are termed “weak D.” Patients with partial D mismatches are at significant risk of sensitization to the D epitopes they lack, but this risk may not be appreciated because patients can type D positive using antisera that recognize intact common D epitopes. Conversely, individuals with weak D variants may express D so weakly as to type D negative with clinical antisera but can still express enough D to be both tolerant to D as recipients and capable of provoking an anti-D alloimmune response in a D-negative recipient. Our understanding of the full range of RH locus variation within the Rh blood group system appears to be incomplete, as supported by a recent detailed study that investigated sickle cell disease patients who developed Rh antibodies despite receiving phenotypically matched transfusions, finding variant RH alleles in 87% of these patients by high-resolution RH genotyping.10 As such, curated up-to-date databases, particularly for partial and weak D alleles, are a priority to inform accurate calling of RH genotypes.
Together, ABO and RH are examples of blood group systems that exhibit the spectrum of genetic and functional variation observed across all blood group systems. ABO harbors relatively simple genetic variation but has hundreds of alleles and a gene that indirectly generates blood group antigens. Conversely, the RH locus directly encodes the proteins presenting DCE antigens, but it is multigenic; the Rh proteins are complex multipass transmembrane domains, and their genes exhibit both simple and complex structural variation. Thus, an approach that can accurately assign ABO and RH blood group genotypes should be capable of calling DNA variants in any blood group gene.
Clinical significance of blood types: allosensitization
Allosensitization is the direct result of exposure to allogenic blood type mismatch(es) and can lead to significant morbidity and mortality from acute HTRs, delayed HTRs (DHTRs), and hemolytic disease of the fetus and newborn, which affects up to 1 in 500 pregnancies.11 Non-ABO-related HTRs are the cause of 15% of transfusion-associated deaths in the United States,12 and nationally the risks of non-ABO HTR are reported to be 1 per 124,525 components transfused. The risk of DHTRs is even more common, at 1 per 20,569 component transfused,12 with high suspicion that these adverse events are under-recognized and underreported. In light of the >13.7 million red cell units that are transfused annually in the United States alone,13 adverse events attributable to allosensitization represent a significant burden. Matching donor units to patients by blood type, when possible, is the only strategy that can avoid provoking alloimmune reactions attributable to existing alloantibodies and prevent primary allosensitization events.12
Extended blood type matching: a strategy to decrease alloimmunization
Extended blood type matching beyond ABO and D has long been proposed as a strategy to reduce alloimmunization.14 Extended matching strategies have been studied in sickle cell disease and implemented in clinical practice for more than a decade. Now there are many institutions matching for Rh C and E and Kell (K) antigens, reserving additional blood type match levels for alloimmunized patients.15-22 This strategy has demonstrated success in reduced alloimmunization rates (to <4% in some reports) in sickle cell disease,15-21 and it is recommended that all sickle cell disease patients undergo extended blood typing early in life.9 Other patients at high risk for allosensitization, particularly multiply transfused patients, such as patients with other hemoglobinopathies23 or those awaiting transplant,24 are also candidates to similarly benefit from extended blood typing strategies.
Blood type testing by serology
Serology, or antibody-based typing, has been the mainstay of blood type testing for >100 years (Figure 3). Several of the most common blood group antigens can be rapidly and relatively inexpensively typed this way. However, the identification of unusual variants of the major common antigen systems and typing of minor blood group systems requires the use of reagents often available only in specialized reference laboratories (Table 1).

Schematic of blood type testing technologies. Illustrated are the progression of increasing information obtained from different blood type testing technologies: serology (left) provides data limited to the targets of the antibody, and molecular testing (middle) provides information on select genetic variants or small genetic regions and can include RFLPs, SSPs, SNPs, and Sanger sequencing. NGS (right) can capture sequence variation at all blood group gene loci at once. Adapted from Johnsen et al39 and Singleton et al.42
Schematic of blood type testing technologies. Illustrated are the progression of increasing information obtained from different blood type testing technologies: serology (left) provides data limited to the targets of the antibody, and molecular testing (middle) provides information on select genetic variants or small genetic regions and can include RFLPs, SSPs, SNPs, and Sanger sequencing. NGS (right) can capture sequence variation at all blood group gene loci at once. Adapted from Johnsen et al39 and Singleton et al.42
Currently, U.S. donors and patients are routinely serologically typed only for ABO and D, and, outside the United States, sometimes K is included in the routine blood type assessment for women of childbearing age. Large-scale serologic testing of donors is time and resource intensive, and only a small percentage of donors and patients are serologically tested for extended blood types (usually E, e, C, c, and K). Individuals harboring other variant blood group antigens are most commonly identified when they allosensitize or during screens of donors for specific antigen-negative units.
In practice, contemporary serologic blood type testing prioritizes prevention of the most common and life-threatening ABO and D transfusion mismatches, followed by a risk-based tiered approach to extended testing. For patients of similar ancestry to the donor population and/or those patients who are expected to have limited transfusion exposures, this tiered serologic approach has been generally successful in delivering well-tolerated transfusions. However, there are still many individuals who become sensitized, ranging from 1% to 2% in the general population25-27 to 20%27 or more10,28 in sickle cell disease. When this occurs, allosensitization almost always occurs in one or more blood group antigens not assessed in routine blood type testing.29
Blood type molecular testing and genotyping
Within the contemporary blood bank laboratory molecular tool kit are variations on older DNA techniques, including single nucleotide polymorphism (SNP)-specific polymerase chain reaction (SSP) and restriction fragment length polymorphism (RFLP), which are used for investigations of less common blood group variants, especially in Rh.10,30 However, these approaches are time intensive and narrow in scope (Figure 3). More recently, SNP panels have been developed to genotype known common and rare blood group SNPs to assign >30 extended blood group antigens,31,32 including the recently Food and Drug Administration-approved Precise-Type HEA Test (Immucor) and the BLOODCHIP platforms (Grifols). SNPs have been useful in providing unprecedented access to extended blood type profiling at scale, particularly for blood group antigens without available testing reagents. However, SNP detection is biased in that it is insensitive to certain types of DNA variation, particularly indels and SVs, and SNP panels can only assess known variants. ABO and RH are not represented on the FDA-approved SNP platform, although other molecular tools providing genetic information in these systems are available commercially, as laboratory developed tests, and several are in development.10,33
With advancements in technologies to enable testing genetic variants at scale, particularly for SNPs, the practice of using genetically informed blood group antigens has begun to be adopted more widely in blood centers and transfusion services.34-36 The benefits to genotyping are numerous: (1) uncommon blood type variants can be screened at once; (2) blood type information can be obtained for antigens for which antisera are unavailable; and (3) weak or variant blood type antigens that confound conventional antisera can be predicted. The main detractors to saturation of genotyping for blood typing have been cost and turnaround time relative to serology. Both of these have decreased markedly in recent years, and, when compared with extended serologic typing or alloantibody identification workups, genetic testing compares favorably.
In blood donors, the scale of genotyping can be used to optimize and manage deep inventories of multiple antigen-negative units, and it has even been proposed that blood group genotypes may be useful as a donor recruiting tool.34 In patients, genotyping is most often implemented for patients at the highest risk for allosensitization or in cases in which allosensitization has already occurred. There is concern that current SNP strategies exhibit ethnic bias. In a recent study of blood type frequencies in Asian and Native Americans, an SNP-based strategy for blood typing was studied and found to have generally high concordance with the 5 blood group antigens tested by serology (C, Jka, Jkb, M, and N).37 However, when considering the presence of SNP-serology discrepancies by individual subject instead of by SNP, 4.5% of all donors exhibited one or more SNP-serologic discrepancies, likely as a result of undetected underlying genetic variation. This highlights that there is value in considering people as a whole, because the likelihood that any one person will harbor a “rare” blood type is combinatorial and much higher than the frequencies of individual rare blood type variants in a population.
DNA sequencing remains the only approach that can capture relatively unbiased genetic variation by virtue of the ability to detect genetic variation whether it is known, rare, or novel. Sanger sequencing remains a well-established method for identifying blood type genetic variants for particularly difficult cases in the transfusion reference laboratory.38 However, the use of Sanger methods for more comprehensive blood group gene locus sequencing is prohibitive in practice because of the size of the regions to be sequenced, low throughput, and relative insensitivity to SVs. With advances in next-generation sequencing (NGS) technology, DNA sequencing can now be performed at the scales needed for more comprehensive and high-resolution blood type genotyping.
Next-generation DNA sequencing
Massively parallel sequencing, also known as NGS, is capable of assessing all types of common and rare DNA variants. The success of NGS is demonstrated by the large number of publications in recent years, and NGS is now implemented for numerous clinical applications, including pharmacogenomics, inherited congenital syndromes, inherited cancer risk genes, and tumor profiling.39 Additionally, whole genome sequencing (WGS) has now been used successfully in the diagnosis of rare, Mendelian disorders,39 and as technology continues to advance, WGS will likely become a mainstay of future DNA sequencing efforts.
NGS can provide high-resolution blood group DNA sequence information that is too cumbersome to obtain by Sanger sequencing and cannot be ascertained by other lower-resolution molecular methods (Figure 3). For most NGS platforms, the accuracy of SNV calling is high,39 whereas indels (small and large), SVs, and copy number variants (CNVs) can sometimes be more problematic. Adjustments to sequencing protocols, such as seeking increased read depth or longer DNA sequence reads, can facilitate detection of CNVs and more complex variants. Statistical and machine learning approaches are now being applied successfully to identify variants and accurately and reproducibly call genotypes.40,41 Sequencing-by-synthesis platforms (Illumina) have led NGS DNA sequencing technologies into the clinical space,39 and this technology is the basis of the NGS blood type sequencing discussed in this chapter. However, other NGS technologies are at various stages of development (for review, see Johnsen et al39 ), which may in the future prove to be useful for blood group gene DNA sequencing, particularly the long-read DNA sequencing technologies.
Recent proof-of-concept NGS efforts are now demonstrating the feasibility of sequencing all blood group genes in a targeted capture DNA sequencing approach (the general method for targeted sequencing is illustrated in Figure 4), as well as WGS. The application of NGS to blood group systems has been proposed by several groups,43-45 and proof-of-principle work sequencing or analyzing NGS blood types supporting feasibility have been reported.45-48 Notably, a whole exome sequencing approach for blood typing would be limited to coding and splice variants, missing noncoding variants that are important for blood group gene expression.49-51

Schematic of one form of targeted NGS. The process starts by randomly cutting genomic DNA (or cDNA) into short fragments (a few hundred base pairs in length); oligonucleotide linkers are added to the fragments to generate a library in vitro; libraries are hybridized to oligonucleotides specific to the targeted genetic region; DNA fragments not interacting with the target are washed away; libraries are introduced into a microscope slide with flow channels containing complementary oligonucleotides on the surfaces of the channel to ones on the libraries, thus allowing hybridization to attach millions of individual molecules to discrete locations on the slide; in situ polymerase chain reaction is performed to copy the individual fragments of the library to enhance sequencing detection; single base extension by a DNA polymerase with all four dye terminators extends the sequence one base, the image of the base extension is captured, and this cycle is repeated 100 times from one end of the molecule and 100 times from the other; DNA sequences are assembled and variants different from the reference(s) identified; and blood group gene variants are annotated using phenotype-genotype databases, population frequencies, and computational tools. Adapted with permission from Johnsen et al.39
Schematic of one form of targeted NGS. The process starts by randomly cutting genomic DNA (or cDNA) into short fragments (a few hundred base pairs in length); oligonucleotide linkers are added to the fragments to generate a library in vitro; libraries are hybridized to oligonucleotides specific to the targeted genetic region; DNA fragments not interacting with the target are washed away; libraries are introduced into a microscope slide with flow channels containing complementary oligonucleotides on the surfaces of the channel to ones on the libraries, thus allowing hybridization to attach millions of individual molecules to discrete locations on the slide; in situ polymerase chain reaction is performed to copy the individual fragments of the library to enhance sequencing detection; single base extension by a DNA polymerase with all four dye terminators extends the sequence one base, the image of the base extension is captured, and this cycle is repeated 100 times from one end of the molecule and 100 times from the other; DNA sequences are assembled and variants different from the reference(s) identified; and blood group gene variants are annotated using phenotype-genotype databases, population frequencies, and computational tools. Adapted with permission from Johnsen et al.39
A targeted blood group gene panel has been developed using the MiSeq platform (Illumina) to capture the blood group genes listed in Table 1 and implemented in a substudy of ∼1100 subjects in a previously studied cohort of Asian and Native American blood donors.37 The blood group gene NGS data was analyzed for blood group gene variants predicted to affect the protein sequence, function, and/or expression level. Blood group gene variants were further annotated as to being previously reported or associated with named blood type alleles and cross-referenced with the donor's corresponding serologic blood type (for ABO, D, C, Jka, Jkb, M, and N) and SNP data (24 SNPs to call 38 blood group antigens).
Analysis of NGS data for ABO DNA variants assigned ABO blood group antigens with high concordance to serologic ABO type (>99%). Sequencing also identified numerous rare ABO subtype alleles, including a blood type A donor harboring ABO c.[261delG(;)502C>G]; the donor was heterozygous for the common O allele and an Aweak allele (Aw06), which likely explains the anti-A antibody detected in this donor in the clinical laboratory.
Targeted sequencing of the RH locus identified serologically D-negative blood donors with >99% sensitivity and specificity. Calls of RHD blood group antigens used a combination of SNV and small indel calls, as well as analysis for whole gene RHD deletion events determined by normalized read depth. The results of this approach in a serologically D-negative subject are shown in Figure 5, demonstrating an individual who is heterozygous for the RHD whole gene deletion (RHD*01N.01) and hemizygous for a gained STOP (c.807T>G, encoding RHD*01N.18). In addition to the successful identification of D-negative donors by NGS, rare RHD weak and partial D variants were also identified, as well as canonical RHCE variants for CcEe and strong evidence for RHD–RHCE hybrid alleles. These data are encouraging in that RH can be sequenced using NGS approaches, particularly in assigning D negative. Additional investigations are warranted to determine the sensitivity and specificity of NGS methods for the spectrum of RH clinical variants, particularly addressing the challenges in using short-read-length NGS data to assign alleles representing RH SVs and RHD–RHCE hybrid alleles.

Example of NGS call in RHD. (Left) Whole gene RHD deletions are identified by relative read depth. Homozygotes for RHD deletions have values near the axis (0), hemizygotes exhibit 50% read depth, and individuals with 2 RHD copies have normal read depth. The dashed box indicates a serologic D-negative donor with one copy of RHD. (Right) Integrated genomics viewer (IGV) screen shot of an RHD variant, c.807T>G, identified in the D-negative donor (dashed box). This variant encodes a gain of STOP that has been reported previously (RHD*01N.18). Thus, this individual has one copy of RHD that harbors a known RHD nonsense mutation, explaining the observed D-negative serologic phenotype.
Example of NGS call in RHD. (Left) Whole gene RHD deletions are identified by relative read depth. Homozygotes for RHD deletions have values near the axis (0), hemizygotes exhibit 50% read depth, and individuals with 2 RHD copies have normal read depth. The dashed box indicates a serologic D-negative donor with one copy of RHD. (Right) Integrated genomics viewer (IGV) screen shot of an RHD variant, c.807T>G, identified in the D-negative donor (dashed box). This variant encodes a gain of STOP that has been reported previously (RHD*01N.18). Thus, this individual has one copy of RHD that harbors a known RHD nonsense mutation, explaining the observed D-negative serologic phenotype.
In addition to these analyses in ABO and Rh, numerous predicted functional blood group gene variants were detected in other blood group genes. For example, in the Kidd group, which harbored the highest rates of SNP-serology discrepancies in the original study37 (>7% in Pacific Islanders), multiple predicted loss-of-function variants were identified that explained multiple cases of Kidd SNP-serology discrepancies (Table 2). Overall, this blood group gene-targeted NGS approach demonstrated high concordance with the available serology and SNPs data, including within genetically challenging blood group systems, identified the underlying genetic cause of multiple cases of SNP-serology discrepancy, and further found numerous rare blood type alleles not able to be identified in the extended blood type serology and SNP approach used previously.

Summary
In the modern genomic era, SNP assays have enabled unprecedented access to extended blood type profiling, whereas targeted high-resolution DNA sequencing approaches to blood typing offers the ability to detect common and rare clinically significant blood group genetic variants to inform high-resolution blood typing. Alloimmunization represents a significant burden for both the individual patient and the healthcare system, and there is mounting evidence that clinically significant uncommon and rare variants confound conventional serologic typing and SNP approaches. Targeted NGS multigene panels have proven clinically useful and cost-effective in practice for a variety of clinical applications.52 Additionally, the continuing rapid advancements of DNA sequencing methods and declines in cost offer the promise of clinical WGS and long DNA read length technologies in the near future. Successes from proof-of-principle work using targeted and WGS to assign blood group antigens further bolsters the achievability of a more comprehensive and unbiased DNA sequencing approach. There remain some logistical challenges in bioinformatics to call complex blood group gene variants and clinically prioritize and annotate blood group gene variants identified through sequencing. Integration of high-resolution blood typing will be most successful through collaborative efforts and maintenance of well-curated databases linked to phenotypes available to the entire community. Application of the recent American College of Medical Genetics (ACMG) guidelines53 for the clinical interpretation of genetic variants is recommended strongly for blood typing, although the vast majority of blood type variants are not inherently pathogenic in the absence of allogenic exposures. One consideration for blood group gene variant reporting is to use both legacy blood group allele (usually ISBT) and Human Genome Variation Society nomenclature qualified by ACMG-like tiers of benign (not conferring a blood group antigen), likely benign (>90% certainty the variant does not result in loss or gain of a blood type antigen), uncertain significance, likely antigenic (>90% certainty the variant causes loss or gain of a blood type antigen), and antigenic (known blood type antigen variant or specific allele-defining variant). Using common language, high-resolution genetic blood type profiles become more accessible for clinical decision making and research applications.
Acknowledgments
I thank Kerry Lannert, Meghan Delaney, Tristan Shaffer, Jason Underwood, Barbara Konkle, Debbie Nickerson, Gayle Teramura, Sam Harris, Shelley Fletcher, and Haley Huston for their many contributions in the field of red cell genomics and discussions that are reflected in this manuscript.
Correspondence
Dr Jill M. Johnsen, Bloodworks NW Research Institute, 1551 Eastlake Ave E, Suite 100, Seattle, WA 98102; Phone: 206-568-2200; Fax: 206-587-6056; e-mail: jjohnsen@uw.edu.
