
Abstract
Hemophagocytic lymphohistiocytosis (HLH) was initially described as an inflammatory condition affecting young children but is increasingly diagnosed in adults. Presenting features such as fever, cytopenias, phagocytosis, elevated ferritin, and increased levels of soluble IL-2 receptor are common in both age groups, but the prevalence of several clinical and biochemical criteria differ between pediatric and adult patients. Specifically, an elevated ferritin level does not have the same specificity for HLH in adults, and many other inflammatory conditions need to be considered in the differential. In contrast to the high incidence of infectious triggers seen in pediatric HLH, HLH in adults is often precipitated by a hematologic malignancy. Malignancy-associated HLH has unique manifestations and a uniformly very poor prognosis. Given these differences, diagnostic scoring systems unique to adult HLH have been proposed, and additional prognostic clinical and immunologic parameters are being explored. Although a genetic predisposition is increasingly found to underlie cases of adult-onset HLH, the mutations are less likely to be bi-allelic and differ slightly from those seen in pediatric cases of familial HLH. The facilitating genetic and environmental factors governing presentation of HLH in adults remain elusive. Understanding the clinical aspects and pathophysiology specific to adults with HLH is necessary to tailor therapies derived in pediatric disease to this under-recognized population.
Learning Objectives
Recognize clinical and laboratory aspects of diagnosis more prevalent in adults with HLH
Appreciate differences and similarities between the triggering events and genetics driving HLH in adults and children
No matter the age at diagnosis, hemophagocytic syndromes such as hemophagocytic lymphohistiocytosis (HLH) represent an unbridled inflammatory condition characterized by fevers, cytopenias, hepatosplenomegaly, and phagocytosis by activated macrophages, facilitated by impaired function of natural killer (NK) cells and/or cytotoxic T lymphocytes (CTLs). Certain biochemical markers such as elevated ferritin, increased soluble Interleukin-2 (IL-2) receptor alpha chain levels (ie, sIL-2R or sCD25), high triglycerides, and low fibrinogen reflect this cytokine-driven, cell-mediated inflammation and aid in its diagnosis.1 As HLH is increasingly recognized in adults, clinical, biochemical, immunologic, and genetic features that characterize adult presentation have become apparent. Adults now comprise up to 40% of cases of HLH, with median age of diagnosis being mid 40s to 50s. Cases are even seen above age 70.2 The incidence may be as high as 1 in 2000 adult admissions at tertiary medical centers.3 Below we address what is common and unique in presentation and diagnostic criteria between adults and children with HLH and how these differences reflect the varied etiologies and genetics in the 2 age groups. Distinctions between “genetic” versus “acquired” HLH can no longer be based on patient age alone, as classical HLH-associated genetic variants are increasingly documented in adults.4
Common aspects of etiology and physiology
HLH has long been recognized not as a single disease but as a common inflammatory endpoint for a variety of conditions. What has classically been referred to as “genetic”, “familial”, or “primary” HLH presents primarily in early childhood and is driven by homozygosity or compound heterozygosity for mutations in genes affecting cytotoxic granule composition, transport and release, such as perforin, MUNC 13–4, syntaxin 11, and syntaxin binding protein 2.5,6 HLH can also manifest in certain immunodeficiency states, including but not limited to Chediak–Higashi syndrome, Griscelli syndrome, and X-linked Lymphoproliferative syndrome. “Acquired” or “secondary” HLH, often defined by the absence of the above genetic scenario, can be seen in all age groups but comprises the vast majority of cases presenting in adulthood, where genetic testing has only recently been pursued. Most episodes of both familial and acquired HLH are triggered by an infectious agent, particularly those of the herpes virus family; an underlying hematologic malignancy may also precipitate the presentation, which is more frequently seen in adults. The inciting event is often not identified.2,7 Until recently, it was not recognized that adults manifesting HLH had any underlying genetic or immunologic defects, but this perception is changing.
In all cases, the symptoms of HLH are driven by high concentrations of inflammatory cytokines, including Interleukin (IL)-1, IL-6, IL-10, IL-12, IL-16, IL-18, tumor necrosis factor (TNF)α, and interferon (IFN)γ resulting in activation of dendritic cells, macrophages, and lymphocytes, particularly NK cells and CD8+ T cells. Impairment of cytotoxic activity hampers elimination of the stimulating agent and impairs down-regulation of the immune response through apoptosis, thus fueling the vicious cytokine cycle that manifests as HLH.8
Clinical presentation of adults with HLH
The 2004 revision of the diagnostic criteria for HLH requires molecular testing consistent with HLH or 5 of 8 clinical or laboratory criteria, namely fever, splenomegaly, cytopenias of at least 2 cell lines, hypertriglyceridemia or hypofibrinogenemia, elevated ferritin, elevated sIL-2R, decreased or absent NK-cell activity, and demonstration of hemophagocytosis in bone marrow, spleen, or lymph nodes.9 Elevated transaminases, bilirubin, LDH, and CSF pleocytosis and/or elevated protein are supportive but not definitive criteria. Other common signs include hyponatremia, edema, rash, hypoalbuminemia, elevated LDH, and other lipid abnormalities. The above criteria were derived solely in children. In the HLH-94 trial of 249 children with HLH, mean age was 2 years (range, 0-15), and a quarter had documented familial disease. Almost all had persistent fevers, hepatomegaly, elevated ferritin, and LDH. Elevated liver enzymes and bilirubin, as well as decreased albumin were common, with most other manifestations being found in <50% of affected children.10
There has been a recent explosion of publications focused specifically on the presentation and etiology of adults with HLH in large cohorts of 50 to >200 patients (summarized in Tables 1 and 2). With regard to clinical presentation, a Chinese study of 103 adult HLH patients found ≥85% presented with high fevers, cytopenias, elevated ferritin, liver dysfunction, hypertriglyceridemia, and bone marrow hemophagocytosis. Eighty percent presented with splenomegaly, 65% with hepatomegaly, 61% with hypofibrinogenemia, 53% with lymphadenopathy, 31% with proteinuria, 25% with skin rash, and 13% with CNS involvement.11 Within the US, Otrock et al12 described 73 adult patients (median age, 51 years; range, 18-82) of whom ≥85% manifested fevers, cytopenias, and elevated ferritin. Splenomegaly was seen in 60%, hypertriglyeridemia in 71%, hypofibrinogenemia in 38%, hemophagocytosis in 77%, elevated sIL-2R in 77%, and abnormal NK cell function in 36.4%.12 Outside of the standard diagnostic criteria, 84% of patients had transaminitis, 92% had hypoalbuminemia, 93% had elevated LDH, and 52% had renal insufficiency. Although not well-defined, 42% of adults in the largest review of adult HLH cases to date were also reported to have pulmonary involvement ranging from cough and dyspnea to respiratory failure.2 In sum, nearly all adult patients shared the constellation of features including fevers, cytopenias, phagocytosis, elevated ferritin, and sIL-2R with lower levels of splenomegaly, hypofibrinogenemia, hypertriglyceridemia, and poor correlation of decreased/absent NK function. The variability in NK cell function has been seen in multiple other studies of adults with HLH.3,13 Notable in 3 studies that presented these data, spanning from 2003 to 2014, 1996 to 2011, and 2006 to 2013, respectively, relatively few patients actually had NK function activity or sIL-2R levels analyzed. NK activity was assayed in only 11 of 73, 5 of 62, and 4 of 50 adults, and sIL-2R levels were requested in 31 of 73, 2 of 62, and 33 of 50, respectively.3,12,13
Clinical characteristics of pediatric versus adult-onset HLH
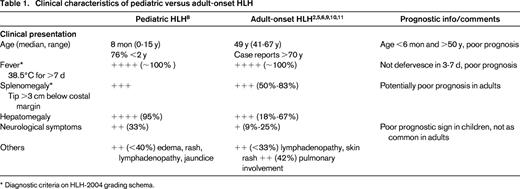
* Diagnostic criteria on HLH-2004 grading schema.
Laboratory characteristics of pediatric versus adult-onset HLH
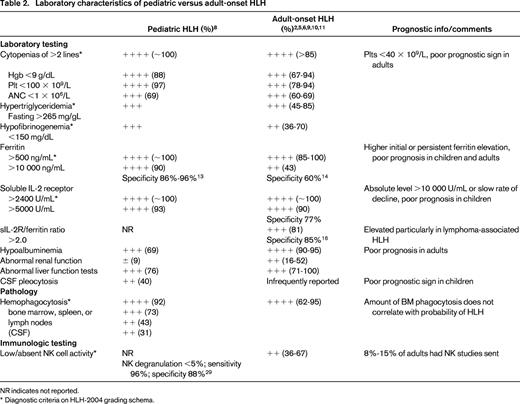
NR indicates not reported.
* Diagnostic criteria on HLH-2004 grading schema.
Although 62% or more bone marrow biopsies were positive for hemophagocytosis among adult studies, the finding of hemophagocytosis did not equate to a diagnosis of HLH. A recent pathologic assessment of bone marrow core and aspirate specimens from 58 patients suspected of having acquired HLH showed that amount of phagocytosis alone did not correlate with probability of having the clinical diagnosis.14
Diagnostic import of ferritin and soluble IL-2 receptor
An extremely elevated ferritin often triggers consideration of HLH in a clinical scenario that could otherwise be interpreted as a severe systemic response to serious infection; conversely a normal ferritin can effectively rule out a diagnosis of HLH. In the original HLH-94 protocol, ferritin of >500 ng/mL had a sensitivity of 84%.10 A serum ferritin level >10 000 ng/mL was reported as 90% sensitive and 96% specific for HLH in a separate pediatric study.15 In a recent comparative study, ferritin >10 000 ng/mL demonstrated a specificity of 86% in children and 60% in adults. In another recent study of 113 adults (median age 58, range, 20-88 years), ferritin levels above 50 000 ng/mL had a specificity of only 17% and a sensitivity of <20% for HLH. Other diagnoses coinciding with high ferritin levels in adults were iron overload (12%), liver failure (11%), sickle cell disease (10%), and graft-versus-host disease (3%) in 1 study and renal failure (65%), hepatocellular injury (54%), infection (46%), myeloid or lymphoid malignancy (32%), rheumatologic/inflammatory conditions (18%), and iron overload (12%) in the other.16,17 A third study indicated a sensitivity in adults of 43% for ferritin >10 000 ng/mL, with 18% of adult HLH cases tested having serum ferritin between 3000 and 10 000 ng/mL.3 In a Japanese study of adult HLH using a lower cutoff of 3000 ng/mL, sensitivity was 66% with a specificity of 68%.18 Thus, HLH should be considered in adults with ferritin levels that are lower than the >10 000 ng/mL threshold typically considered sensitive and specific in pediatric cases, and a wide set of differential diagnoses need to be considered in adults with markedly elevated ferritin levels. The negative predictive value of a normal ferritin, however, remains extremely high in adults as well as children.
In children, a sIL-2R level >2 age-adjusted standard deviations above the mean (typically 2400 U/mL) is reported to be 93% sensitive for HLH, more sensitive than elevated ferritin.9 Unfortunately elevated sIL-2R can also be seen in autoimmune diseases, malignancies, infections (particularly HIV and TB), cardiac damage, and allograft rejection after organ transplantation. The sensitivity and specificity of elevated soluble IL-2R levels in adults has not been as extensively studied. Tabata et al found among 110 adult cases that sIL-2R ≥5000 U/mL, was 90% sensitive and 77% specific for HLH.18 In adults, a sIL-2R/ferritin ratio >2.0 had a sensitivity of 81% and specificity of 85% for HLH. In addition, each measurement was highly correlated with a diagnosis of lymphoma-associated HLH. A higher mean sIL-2R (13.5 vs 4.2; p < 0.005), lower mean ferritin (2561 vs 20 462; p < 0.005), and higher sIL-2R/ferritin ratio (8.6 vs 0.7; p < 0.0005) were seen in lymphoma-associated disease compared to HLH without malignancy.19 In children, absolute ferritin and sIL-2R levels, as well as rate of decline of ferritin, have prognostic implications. A ferritin decrease of <50% has an odds ratio for death of 17 compared to a ferritin decrease of >95%.20 Whether these data hold similar prognostic information in adults has not been studied.
Etiology of HLH in adults
An extensive review of 2197 cases of adult HLH identified that 50.5% of cases were triggered by infection (predominantly EBV and HIV), 48% arose in the setting of malignancy, 44% were in the setting of a lymphoma (split fairly evenly between NK/T cell and B cell lymphomas), and 13% were in the setting of autoimmune diseases, primarily SLE.2 A study of 567 Japanese cases of HLH found 44% of patients were >15 years of age; 19% were adults >60. Genetically-driven FHLH occurred almost exclusively in infants (<1 year old) and autoimmune disease and infection were the most common initiating factors in children ages 1-15. In patients >15 years old, 34% of cases were associated with infection and 34% with malignancies. Lymphoma-associated HLH was found solely in patients >30 years old, particularly in those ≥60 years old.7 Among 162 French adults, 57% of patients had malignancies (primarily B-cell related), 25% had infectious triggers. In this study, 45% of patients were either HIV-positive or on immunosuppressive therapy.21 Multiple other studies have confirmed this distribution in adults with HLH, with 29%-52% of cases coincident with malignancy, 23%- 41% being driven by infection, and 8%-20% arising in the setting of autoimmune or rheumatologic conditions.3,11,13 The most common malignancies were B-cell lymphomas, Hodgkin's lymphoma, and T-cell lymphomas including NK/T-cell subtypes. Regardless of age of onset, malignancy-associated HLH has a uniformly poor prognosis.
In all 6 studies referenced above, the most common infectious trigger was EBV, with markedly higher percentages of EBV infections seen in Asian and, particularly, Japanese groups. Higher levels of circulating EBV DNA during active disease were associated with poor outcomes.22 Although the mechanism of EBV-related HLH has not been definitively established in vivo, expression of latent membrane protein-1, whereby EBV activates NF-kB, upregulates Th1-type cytokines, and leads to T-cell activation in EBV-infected T cells, is postulated to fuel HLH.23 Shortly after the severe 2009 H1N1 influenza A epidemic, HLH was also noted to complicate up to 36% of cases of severe H1N1 infection.24
HLH complicating hematologic malignancies
It is estimated that HLH affects 0.9% of all adults with hematologic malignancies and up to 20% of patients with certain B cell lymphomas, NK/T cell nasal lymphomas, and panniculitis-like T-cell lymphomas.25 Incidence of hemophagocytosis in malignancy seems to be differentially impacted by genetic modifiers present in different ethnic groups. A study of 50 Western patients with intravascular lymphoma revealed no associated HLH, whereas among 123 East Asian and Japanese patients, 19% and 44% manifested HLH, respectively.26 In a study of 1239 Japanese patients with non-Hodgkin's lymphoma, the cumulative incidence rate of HLH was 2.8%. Those patients with HLH had inferior overall survival compared to their peers without HLH (p < 0.0001). The rate of early death within 4 months of developing HLH was significantly higher in patients with NK/T-cell lymphoma than in those with B cell lymphomas (62.5% vs 10.5%).27 Sometimes HLH will manifest during induction chemotherapy for malignancy, particularly in acute leukemias.28
Several groups have identified biochemical features of adult HLH that correlate with underlying malignancy and thus indicate need for further diagnostic investigation. These include lower fibrinogen <1.5 g/L, LDH ≥1000 U/L, an elevated sIL-2R/ferritin ratio, and in several studies platelets <40 × 109/L.29 Conversely, hematologic malignancies can often present with fevers, cytopenias, organomegaly, and elevation of inflammatory markers, so distinguishing concurrent HLH can be a challenge. Novel cytokine assays for inducible protein 10/CXCL10 and monokine-induced by IFNγ/CXCL9 have been piloted to identify when HLH is present with lymphoma, to distinguish lymphoma-associated HLH from sepsis, and to classify severe versus moderate/mild lymphoma-associated HLH. Elevated levels of these 2 cytokines had a sensitivity and specificity of 100% and 95%, respectively, for distinguishing a diagnosis of lymphoma-associated HLH from sepsis and significantly decreased with successful treatment, indicating utility in diagnosis and monitoring disease response.30
Diagnostic criteria for adults with HLH
Because acquired or reactive HLH in adults can be clinically difficult to distinguish from severe sepsis or hematologic malignancy and has attributes distinct from pediatric presentations, metrics for diagnosis specifically in adults have been sought. An expert panel Delphi analysis of 26 criteria revealed positive consensus on 9 “absolutely required” or “important” diagnostic criteria in adults.31 These were unilineage, bilineage, or trilineage cytopenias, hemophagocytosis, elevated ferritin, fever, organomegaly, underlying predisposing disease, and elevated LDH. Of interest, criteria determined to be of “minor interest” or “useless” included rash, liver function test abnormalities, serum albumin, NK cell activity, sIL-2R, and soluble CD163. Building on this, an “HScore” system was recently developed and validated.32 Clinical and laboratory criteria from 162 adults ultimately diagnosed with HLH and 104 adults without HLH were reviewed and subjected to logistic regression analysis. Three clinical variables (underlying immunosuppression, high temperature, and organomegaly) were combined with 5 biologic variables (triglyceride, ferritin, liver SGOT/ALT, fibrinogen, and cytopenias) and 1 cytologic variable (hemophagocytosis on bone marrow aspirate) and weighted. The probability of having HLH was <1% for an HScore of ≤90 and >99% for an HScore of ≥250.
Immunologic phenotypes in HLH
In familial HLH, severe impairments in cytotoxic granule composition, trafficking, and release are seen even in the presence of normal NK cell numbers; patients with familial HLH typically manifest persistent decreased or absent NK or CTL function. NK cell defects have been characterized by cytotoxicity assays such as chromium release or flow cytometry-based assays measuring intracellular perforin and granzyme B expression; CD107a mobilization has also been followed as a marker of degranulation. Resting NK cell degranulation assays of <5% reportedly have a sensitivity of 96% and specificity of 88% for familial HLH in children.33 In acquired HLH, patients may have low NK cell numbers and decreased NK function during periods of disease activity that are thought to revert to normal after treatment. In one experience, 20 of 30 critically ill patients with HLH manifested significantly decreased NK cell activity, but in other studies 50% or fewer adults demonstrated decreased NK activity when assayed.12,34 There have been attempts to standardize these assays in adults, but interpretation of values remains difficult. Additional testing of the effects of IL-15 and blocking of Fas ligand on NK and CTL activity is reported to differentiate between primary and secondary HLH.34 However, the finding of normal NK function, particularly during active symptomatic disease, should not preclude the diagnosis of HLH. As noted above, testing of NK cell function in adults is not widely pursued as its significance in diagnosis and management remains unclear.
Although the immunologic stimuli driving acquired HLH seem myriad, a plausible underlying hypothesis is that chronic antigen stimulation by a viral pathogen or malignancy may unmask a predisposition to T- and NK-cell dysfunction. Therefore, it makes sense in adults to augment NK cell testing with other markers for lymphocyte activation and dysregulation. These include levels of soluble CD163, which is a “scavenging” receptor for hemoglobin-haptoglobin complexes and a very specific marker of macrophage activation, and quantification of CD14+CD16bright monocytes. A combination of sIL-2R and sCD163 testing has been piloted to identify subclinical presentations of HLH complicating rheumatologic conditions.35 Levels of IFNγ and IL-10 have also been investigated as diagnostic and prognostic tools for distinguishing HLH from sepsis and acute viral infections. Levels of IFNy >75 pg/mL and IL-10 >60 pg/mL have 99% sensitivity and 93% specificity for HLH, compared to isolated elevations of IL-6 as are seen in sepsis.36 Patients with secondary HLH also have severe imbalances of IL-18 to IL-18 binding protein ratios versus normal controls, perhaps enabling Th1 lymphocyte and macrophage activation.37 Decreased CD3+ and CD8+ T cells numbers and lower CD4:CD8 ratios are associated with inferior survival in HLH.38 Abnormal T-cell profiles, such as loss of CD5, CD7, and/or CD3 have been noted in EBV-associated HLH.39 How this immunologic testing should be incorporated into diagnostic algorithms, how results differ with age of onset and etiology of HLH, and how findings correlate with outcomes remain unknown.
Predictors and outcome in the 2 age groups
Although outcomes are variable within subgroups based on underlying etiology, children with HLH have historically fared much better than adults, with the standard treatment algorithm being the HLH-94 or HLH-2004 protocols consisting of pulses of etoposide and dexamethasone followed by calcineurin inhibition. For those with recurrent, refractory, or familial HLH, hematopoietic stem cell transplant (HSCT) is the therapy of choice, ideally from a fully-HLA matched donor. In the HLH-94 pediatric study, 5 year survival was 54%; 3 year survival was 64% for those able to undergo HSCT.10 Higher elevations of bilirubin, increased CSF cell counts at diagnosis, thrombocytopenia, initial and persistent ferritin >2000 ng/mL, or continued fever 2 weeks into therapy predicted for decreased survival.40 Alternative induction therapy utilizing anti-thymocyte globin along with steroids and calcineurin inhibition have also been used successfully. In a smaller single-center study, active disease at time of transplant and use of a haploidentical donor correlated with decreased survival.41
Most studies of adults to date show inferior outcomes, particularly for those with malignancy-associated HLH. Among 1109 adult cases collated from numerous publications, the mortality rate was 41%.2 In the large Japanese study, 5 year OS was >80% for patients with EBV, or other infection, and autoimmune-associated HLH, 48%-54% for familial HLH or B-cell lymphoma-associated HLH, and 12% for those with NK/T-cell lymphoma-associated HLH.7 Increasing age had an independent negative impact on survival within EBV- or B-cell lymphoma-associated HLH. Consistent findings in other studies were increased early mortality in patients with underlying malignancy (eg, median OS 1.1 vs 47 months and 1.4 vs 23 months).3,12 Other consistently cited adverse prognostic factors were increasing age and decreased platelet count <40 × 109/L.21 Occasionally cited adverse factors were male sex, presentation with splenomegaly, active EBV infection, fever not subsiding within 3 days of diagnosis, occurrence of DIC, hypoalbuminemia, and lack of etoposide during management.42 Ferritin levels >50 000 ng/mL were a predictor for 30 day mortality (HR 3.3).3 Unfortunately, although prognostic factors relevant to adults are now recognized, systematic analyses of efficacy for standard and novel therapies in adults are still lacking.
Genetic determination of presentation
Classic familial HLH presenting in early childhood has been grouped into 5 genetic subtypes based on underlying genetic defects inherited in an autosomal recessive fashion: namely FHLH-1 9q21.3-locus 56; FHLH-2 PFR1; FHLH-3 UNC13D; FHLH-4 STX11; and FHLH-5 STXBP2/UNC18B. The gene mutated in FHLH-1 has not yet been identified. The other genes associated with FHLH play a role in cytotoxic pore formation, cytolytic granule secretion, intracellular vesicle transport, and membrane fusion, respectively. Children with mutations in the same gene can manifest different clinical presentations. As an example, FHLH-2 with canonical perforin mutations is typically characterized by early onset (<2 months of age), very high ferritin and sIL-2R levels, and persistent deficiencies in NK cell activity.43 However, children with missense mutations in PRF1 develop disease later and show only moderately reduced cytotoxicity. Similarly, patients with splice-site mutations in STXBP2 develop clinical manifestations at an older age versus those with nonsense mutations (median, 4.1 years vs 2 months).6 Clinical manifestations may also be dictated by the particular combination of autosomal recessive mutations. Patients who are compound heterozygotes for 2 mutations in the degranulation pathway (UNC13D, STXBP2, STX11, and RAB27A-Griscelli Syndrome) have early age of onset similar to that of patients homozygous for biallelic mutations. Those with a single mutation or with compound heterozygosity for a perforin and degranulation pathway mutation manifest disease later.44 Thus, the type and combination of mutations in FHLH seems to correlate with age of onset, symptoms, and magnitude of cytolytic impairment in children.
This phenomenon may explain the frequency with which homozygous, compound heterozygous or single mutations/variations in classical FHLH genes are being found in symptomatic and asymptomatic adults as genetic testing becomes more widely available. Hypomorphic missense and splice-site variants in perforin (PRF1), syntaxin 11 (STX11), and syntaxin binding protein 2 (STXBP2) genes have been found in single heterozygous, compound heterozygous, and homozygous states in up to 14% of 175 adults referred for genetic testing related to a suspected diagnosis of HLH. This is compared to a mutational frequency of 30% in children <18 years referred for the same testing, in whom nonsense and null mutations resulting in loss of protein expression were more commonly seen.4 Not all those referred for testing would have been diagnosed with acquired or familial HLH, making the frequency with which mutations were seen in adults even more surprising. These findings support routine genetic testing even in older patients with HLH and calls into question the concept of “familial” or “genetic” HLH as being primarily a childhood disease. Among 252 Chinese adolescents and adults with HLH, mutations in 6 HLH-associated genes were found in 7.1% of individuals, with changes in PRF1 being most common.45 A separate group found that 7 of 8 patients with late-onset FHLH carried at least one splice-site mutation in UNC13D or STXBP2, in a homozygous or compound heterozygous state. In these patients, NK cell cytotoxicity, and NK and CTL degranulation were impaired, but CTL function was normal. NK cell degranulation could be partially reconstituted by IL-2 and CTL cytotoxicity in vitro was normal: a phenotype distinct from patients with early-onset FHLH.46
More than one-half of the 25 adults found by Zhang et al to have hypomorphic sequence variations had only one variant in any of these genes; the majority being in perforin, followed by MUNC13-4 and syntaxin binding protein 2.4 In silico analyses of the predicted structural effects of the missense variants ranged from “benign” to “probably damaging”. The best studied of such single nucleotide polymorphisms (SNPs) is the A91V PRF1 variant which is present in >4%-7% of the general healthy population, and yet has been correlated with absent or low levels of perforin expression and variability in cytotoxic function in individuals who are heterozygous or homozygous for this SNP.47 Heterozygosity for the A91V variant is enriched in patients with HLH. In 1 study of clinical FHLH, 10 of the 24 patients with heterozygosity for A91V had additional mutations in either MUNC13-4 or perforin. Thus, A91V PRF1 mutations may be permissive but not sufficient for HLH.48 In those with hypomorphic sequence variations, NK cell function was not closely correlated with the number of SNPs or variations present in PRF1, UNC13D, or STXBP2 in a given individual.4 How many of these individuals, particularly those with single variants, actually manifest clinical HLH or have a familial syndrome, and how many individuals harboring the same SNPs will never develop HLH cannot be answered by these studies. Large genomic databases derived from healthy adults and those with various diseases may allow us to address in the future what risks are posed by individual polymorphisms in classic FHLH genes and what other genes contribute to the manifestation of clinical disease.
Last, variants in some of the classical FHLH genes may drive other hematologic and inflammatory diseases. Biallelic perforin SNPs have been found in 4 of 29 patients with Hodgkin and non-Hodgkin lymphoma.49 Heterozygosity for specific variants within the perforin gene have been suggested as a susceptibility factor for lymphocytic leukemias, childhood anaplastic large cell lymphoma, autoimmune lymphoproliferative syndrome, as well as type I diabetes and multiple sclerosis.50 The evolving genetic data in adults now frame HLH not as a binary syndrome of familial versus acquired disease, but as a continuum across all age groups with differential disease penetrance and presentation being driven by some shared and some unique genetic drivers interacting with multiple as yet unknown modifiers.
Next steps
Although children and adults share many features in the clinical presentation and underlying pathophysiology of HLH, there are differences between the age groups, particularly in biochemical profiles, underlying genetics, and inciting etiologies. The increased incidence and abysmal prognosis of malignancy-associated HLH, as well as the large differential diagnosis of elevated ferritin and inflammatory symptoms in adults, mandates further investigation into age-specific diagnostic systems, such as the HScore. Presentation with missense mutations in a single allele of genes typically associated with familial HLH suggests additional predisposing factors for those presenting with sporadic cases in adulthood. Increased awareness and diagnosis of HLH in adults will enable larger studies within this age group to further delineate prognostic variables and, most importantly, clarify optimal treatment regimens. These may or may not include the HLH-2004 regimen and stem cell transplantation; although the evidence of significant genetic overlap between pediatric “familial” and adult “acquired” HLH may indicate success for a common therapeutic approach. As an example of the dilemmas associated with managing adults with HLH, consider the case of a 50-year-old male presenting with an isolated missense single nucleotide polymorphism in STXBP2, whose HLH responded well with rapid normalization of ferritin after initial etoposide and immunosuppressive therapy. His only potential fully matched stem cell donor is an asymptomatic 52-year-old sibling carrying the same STXBP2 variant. Characterization of this patient's disease as “genetic” versus “acquired”, his prognosis and optimal course of therapy, are currently undefined. Cases like this are increasingly common and highlight why a better understanding of what distinguishes onset of disease later in life is crucial for adults presenting with HLH.
Correspondence
Nancy Berliner, Division of Hematology, Brigham and Women's Hospital, 75 Francis Street, Boston, MA 02115; Phone: 617-732-5840; Fax: 617-264-5215; e-mail: nberliner@partners.org.
