
Abstract
The hemostatic balance changes with advancing age which may be due to factors such as platelet activation, increase of certain clotting factor proteins, slowing of the fibrinolytic system, and modification of the endothelium and blood flow. Generally, this predisposes the elderly to thrombosis rather than bleeding. It often necessitates antiplatelet or anticoagulation therapy, which can cause significant bleeding problems in an aging population. Additionally, changing renal function, modification in immune regulation, and a multitude of other disease processes, can give rise to acquired bleeding disorders. Bleeding can prove difficult to treat in a dynamic environment and in a population that may have underlying thrombotic risk factors.
This article discusses some specific challenges of acquired bleeding arising in the elderly. The use of anticoagulation and nonsteroidal anti-inflammatory medications is prevalent in the treatment of the elderly and predisposes them to increased bleeding risk as their physiology changes. When prescribing and monitoring these therapies, it is exceedingly important to weigh thrombotic versus bleeding risks. There are additional rare acquired bleeding disorders that predominantly affect the elderly. One of them is acquired hemophilia, which is an autoimmune disorder arising from antibodies against factor VIII. The treatment challenge rests in the use of hemostatic agents in a population that is already at increased risk for thrombotic complications. Another rare disorder of intensifying interest, acquired von Willebrand syndrome, has a multitude of etiologic mechanisms. Understanding the underlying pathophysiology is essential in making a treatment decision for this disorder.
Learning Objectives
To recognize that coagulation changes with age and that it may predispose elderly patients on antithrombotic therapies to bleeding
To recognize the underlying thrombotic risk and comorbidities complicating the treatment of acquired hemophilia A
To understand the various pathophysiologic mechanisms leading to Acquired Von Willebrand Syndrome and assign appropriate treatment according to that mechanism
Acquired bleeding disorders in the aging population are of concern because they are not readily diagnosed. In a study, community physicians from various specialties were presented with a hypothetical older adult woman complaining of recurrent epistaxis. Although most practitioners ordered complete blood counts and activated partial thromboplastin time (aPTT) as part of their work-up, less than one-half of nonhematologists repeated a significantly prolonged aPTT and <45% consulted a hematologist. Of those, emergency medicine physicians were least likely to consult hematology.1
The reasons why bleeding disorders in the aging are overlooked could be due to the fact that aging is usually associated with thrombotic rather than hemorrhagic complications. Early signs of a bleeding disorder such as bruising are easily dismissed as “senile purpura” or “normal” side effects of drugs they are taking such as antiplatelet agents and anticoagulants.
Hemostasis changes with aging as a result of change in coagulation factors, thrombin generation, and platelet function. Platelet activation and most coagulation factors (fibrinogen, factor V, VII, VIII, IX, XI, XIII, and VWF) increase with age. Several studies have shown that this translates into increased thrombin generation.2 At the same time, fibrinolytic activity slows with age.3 Besides these dynamics, aging is associated with change in endothelium, blood flow through the vasculature and impaired renal function, all affecting how hemostatic agents interact and ultimately influencing coagulation.2 Clinically, the risk for arterial and venous thrombosis increases with age2 In the United States, 70% of people >60 years and >80% of those >80 years have hypertension, dyslipidemia, and/or other forms of coronary artery disease.4 Similarly, malignancy is associated with an increased risk for venous thrombosis and predominantly affects older individuals, with 75% of cases diagnosed in persons aged ≥55 years. Trousseau reported that localized cancer can induce hypercoagulability in the 1860s and it was subsequently recognized that many types of tumor cells express tissue factor and/or other clotting initiators.
In the elderly population, development of an acquired bleeding disorder poses a challenge because it presents when thrombotic risk factors are present, but also in the setting of other comorbidities. The new onset of bruising and bleeding symptoms could be the proverbial canary in the mine and be the first indication of an evolving underlying disease. This could be from thrombocytopenia associated with a liquid or solid malignancy or immune thrombocytopenia purpura (ITP). It could also be related to disseminated intravascular coagulation (DIC) due to malignancy or sepsis. Lymphoproliferative disorders have been associated with acquired bleeding disorders, such as ITP, acquired hemophilia, acquired von Willebrand syndrome, and less commonly thrombasthenia, acquired factor X-, V-, XI-, XII-, or prothrombin deficiency.5
In an elderly person who develops an acquired bleeding disorder and possibly needs hemostatic intervention, there can be a delicate tightrope between bleeding and clotting. This article is not intended to be a comprehensive review of all bleeding disorders that can surface in the aging population, but rather discuss some of the challenges that could arise.
Drug induced
Bleeding in an elderly patient is commonly iatrogenic. More than 2 million people in North America are treated with oral anticoagulation and many more are taking antiplatelet agents, nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin. They all carry an inherent bleeding risk, which increases with aging. Chronic pain escalates with age and so does the use of NSAIDs and aspirin. Atrial fibrillation (A-fib) is common in the elderly and the largest indication for anticoagulation so as to prevent stoke and systemic embolism. Additionally, the risk for cardiovascular disease and venous thromboembolism increases in the elderly and often necessitate the use of one or more antithrombotic agents, which leads to a significant increase in bleeding risk.6 Patients should continuously be monitored for how they are actually taking the medications, a change in their renal function and any bleeding symptoms.
Bleeding risk on anticoagulation.
The incidence of A-fib increases with age and affects ∼9% of people >80 years of age. At the same time, thrombotic complications associated with A-fib increase with age. Per the commonly used CHA2DS2-VASc score, age >75 years alone warrants anticoagulation with an oral anticoagulant, such as warfarin, dabigatran, rivaroxaban, apixaban, or edoxaban7 to prevent thrombotic complications. At the same time, people >65 on oral anticoagulation for stroke prevention are at significantly increased risk for major bleeds, 15%-20% being intracranial hemorrhages which are associated with significant mortality (46%) and severe disability (22%).7 As people age, renal function tends to decline. In people with very poor renal function (eGFR <15 mL/min/1.73 m2), the incidence of major bleeding is more than 10-fold higher than in people with normal renal function (≥90 mL/ min/1.73 m2) during the first 30 days of treatment with warfarin. The risk of major bleeding can be estimated by the HAS-BLED bleeding risk score which is increased in the presence of hypertension, abnormal renal/liver function, history of stroke, bleeding history, or predisposition, labile international normalized ratio (INR in range <60% of time), age >65, and alcohol of >8 drinks a week.7
A meta-analysis of available phase III data through 2013 indicated better efficacy of targeted oral anticoagulants (DOACs; direct thrombin inhibitor: dabigatran, and anti Xa inhibitors: rivaroxaban, apixaban) over warfarin in preventing thromboembolism in A-fib and deep venous thrombosis recurrence in people >75 years. It further showed that the risk of major or clinically significant bleeding was not more for the DOACS over warfarin.8 In another meta-analysis comparing bleeding rates between vitamin K agonists and DOACs, it appears that fatal bleeding and intracranial hemorrhage may be reduced when using DOACs, especially in the elderly.9 Of particular note, impaired renal function and advanced patient age were found to be independent risk factors for bleeding complications in patients taking dabigatran.10
Treatment of anticoagulation induced bleeding
For an elderly person on anticoagulation, the best approach is prevention by frequent monitoring for bleeding, medication compliance and renal function. Interestingly, a recent Swiss study showed that increased physical activity was protective of major bleeding on anticoagulation.11 For the treatment of warfarin associated acute bleeding, 4-factor prothrombin complex concentrate (4F-PCC, containing clotting factors II, VII, IX, and X) was superior to fresh frozen plasma (FFP) in reducing INR and non-inferior in controlling bleeding (bleeding control was 72.4% with 4F-PCC and 65.4% with FFP).12 For patients needing urgent surgical or invasive interventions, 4F-PCC appears to be superior to FFP for reduction of INR (55% vs 10%, p < 0.0001) and in achieving hemostatic control (90% vs 75%, p = 0.0142) without increase in thromboembolic events.13 The reversal of direct oral factor Xa inhibitors (rivaroxaban, apixaban, edoxaban, betrixaban), and the direct thrombin inhibitor (dabigatran) is still problematic and under investigation. FFP, PCCs, and recombinant factor VIIa have been investigated with limited utility. Dabigatran can be removed via dialysis and a Fab fragment of a monoclonal antibody (idarucizumab) is awaiting approval by the FDA as a targeted reversal agent. Concurrently, a modified recombinant FXa (andexanet alpha) is being developed as an antidote for direct FXa inhibitors.14
Renal impairment
Approximately 13.6% of adults in the US have chronic kidney disease according to the National Health and Nutrition Examination Survey (NHANES) and the prevalence is significantly higher in people >60 years of age. The bleeding diathesis associated with renal failure is largely explained by a functional platelet disorder.15 Thus, although tests of primary hemostasis (bleeding time, platelet function analyzer) could be abnormal, tests for secondary hemostasis (prothrombin time, activated partial thromboplastin time) will remain normal. The platelet dysfunction can be explained by a uremic effect on the platelet, as well as associated anemia with lack of sufficient cellular mass within the vasculature to push platelets to the periphery where they aggregate and activate.
Although bleeding symptoms associated with renal impairment may manifest as increased bruising, chronic kidney disease is also associated with increased perioperative bleeding.16 The degree of bleeding inversely correlates with the degree of anemia and correction of the hematocrit to ∼30% (with transfusion and/or erythropoietin) is helpful in controlling bleeding symptoms.17
Acute bleeding can be managed with dialysis to correct the uremia, transfusion of red blood cells to correct the anemia, and desmopressin (0.3 mg/kg IV in 50 mL saline >30 min or intranasally at the same dose). Desmopressin will work for several hours and has decreased effect with repeated dosing. Conjugated estrogens (0.6 mg/kg IV daily for 5 days) can be helpful to treat chronic bleeding.15
Acquired hemophilia A (AH)
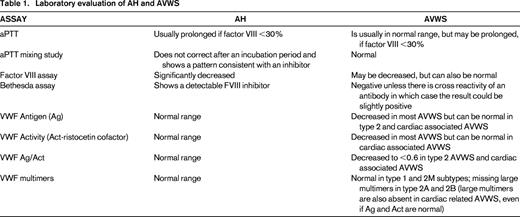
Acquired hemophilia A is a very rare bleeding disorder affecting ∼1.5 per million annually18 and is caused by the development of IgG auto-antibodies to endogenous factor VIII (FVIII). Though it can be seen in women associated with pregnancy, it mainly affects the aging population (median age at diagnosis 73.9 years).19 In contrast to congenital hemophilia, both sexes can be affected; most patients (89%) are diagnosed after investigation for a bleeding event, but sometimes a prolonged aPTT without symptoms triggers the work-up. Diagnostic laboratory evaluation for AH is summarized in Table 1. Bleeding symptoms are usually spontaneous, severe, and cutaneous, mucosal or muscular in nature.19 The incidence of AH rises with age. Although it can be idiopathic, it is associated with an underlying medical condition in ∼50% of cases. The main underlying conditions are malignancy or autoimmune disease, but others include underlying infection, monoclonal gammopathy of undetermined significance, dermatologic conditions, and the use of certain drugs (eg, antibiotics, clopidogrel, interferon). Whether there is an underlying association or not, AH in the elderly can occur in the setting of thrombotic risks, such as previous venous thromboembolism, coronary artery disease, and malignancy.
Challenges in treating bleeding in AH.
A challenge in treating AH in the older population is that it can occur in a prothrombotic background. AH tends to present with severe bleeding symptoms requiring hemostatic agents. The two commonly used bypassing agents, recombinant factor VIIa (rFVIIa, ∼90 mcg/kg every 3 hours) and activated prothrombin complex concentrate (aPCC, ∼67 U/kg every 12 hours) have been shown to be effective agents for bleeding control and will achieve effective hemostasis in >90% of bleeds when given as first-line agent.20 Both agents do not have a surrogate laboratory measurement to guide treatment. While hemostasis can often be assessed clinically, supra-therapeutic ranges are difficult to predict and can predispose the patient to thrombosis. Reports of thrombotic complications with the use of these agents between 2.9% and 8.6% have been reported,20,21 and both arterial and venous events have been described. Teasing out the true incidence and causative nature is difficult because most reports and studies were not set up and did not have the follow-up required to answer the question of thrombotic risk. In addition, it is impossible to gage how much the addition of the bypassing agents raised the already existing risk for thrombosis in this population. Utilizing treatments that could be monitored with laboratory tests, is therefore of interest and may lessen the risk of unwanted thrombosis. Although both human FVIII concentrates (exogenously) and desmopressin (DDAVP, endogenously) can increase measurable FVIII levels, they are both less effective treatments in AH due to inactivation of human FVIII by the circulating antibodies. Porcine FVIII has enough similarity to human FVIII to be of hemostatic value and achieve measurable FVIII levels in human blood. Yet, it is different enough to potentially escape the unwanted immune response, at least temporarily. Plasma derived porcine FVIII was successfully used for the treatment of AH22 between 1984 and 2004, but then taken off the market due to potential contamination with porcine parvovirus. In October 2014, a recombinant porcine FVIII (rpFVIII) was licensed in the US for the treatment of AH. In a phase III clinical trial, it was 100% effective at controlling serious life or limb threatening bleeding within the first 24 hours and overall controlled bleeding in 86%.23 At initiation of treatment with rpFVIII, 10/29 (34%) subjects had cross reactive anti-porcine FVIII inhibitors (0.8-29 BU) and needed rapid repeat dosing after the initial dose, but all achieved measurable FVIII levels of >100% within the first 24 hours. The initial response to rpFVIII at 200 U/kg varied significantly with recovery levels between 20% and 775% and was dependent on the underlying porcine inhibitor level. Routine baseline one-stage factor VIII activity testing gave reliable results and redosing could be tailored. Five subjects developed de novo anti-pFVIII antibodies 8, 18, 22, 35, and 85 days after the initial rpFVIII infusion, rendering treatment ineffective in some of them. There were no thrombotic complications reported with the use of rpFVIII in this clinical trial, but the overall observational period was limited to 90 days.23
Bypassing agents and treatment with human or porcine FVIII concentrate is expensive and cost has to be taken under consideration, especially when high and frequent dosing is required. Treatments for bleeding in AH are summarized in Table 2.
Treatment of bleeding in AH
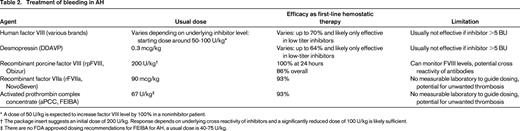
* A dose of 50 U/kg is expected to increase factor VIII level by 100% in a noninhibitor patient.
† The package insert suggests an initial dose of 200 U/kg. Response depends on underlying cross reactivity of inhibitors and a significantly reduced dose of 100 U/kg is likely sufficient.
‡ There are no FDA approved dosing recommendations for FEIBA for AH, a usual dose is 40-75 U/kg.
Challenges with immunosuppression in AH.
The standard approach to eradicate the antibody (inhibitor) in AH is immunosuppressive therapy (IST). Inhibitors rarely disappear spontaneously and failure to eliminate the inhibitor is associated with higher risk of mortality.21,24 Corticosteroids used with cyclophosphamide have the highest rate of stable complete remission (70%), closely followed by rituximab in combinations with steroids, cyclophosphamide or other immunosuppressive agents (63%-67%).21 Predictors of IST success and overall better outcomes are higher baseline FVIII levels, lower inhibitor titers, the absence of malignancy and an overall better performance status.21,25 Though it is clear that prompt initiation of immunosuppressants is indicated in the treatment of AH, it comes at a considerable cost of infectious complications and sepsis. The rate of complications from IST is ∼30%25 and mortality risk form IST complications is estimated as high as 23%.24 The combination of immunosuppressants leads to quicker and increased stable response rates (48% for steroids alone, 42% for rituximab alone vs 70% for steroids/cyclophosphamide, 64% for steroids/rituximab).21 However the risk for infection rises as immunosuppressants are combined (16% for steroids alone vs 27% for steroids/cyclophosphamide). Health care associated infections linearly rise with age26 and infection in the elderly poses an increased mortality risk; thus, any additional risk factors exposing the elderly to infections have to be of particular consideration and the risk of mortality from bleeding vs. infection has to the weighed in the treatment decision.
Acquired von Willebrand syndrome (AVWS)
Acquired von Willebrand syndrome is exceedingly rare. It has however gained increasing interest, especially in association with cardiac conditions.27,28 It was first described in 1968 before the availability of standardized von Willebrand factor (VWF) testing. Because there are no large prospective studies, it is difficult to assess the actual prevalence, but it is likely underreported due to lack of awareness of this disorder in the community, especially in the elderly population where milder bleeding symptoms are often disregarded. Unlike acquired hemophilia, which has a distinct pathophysiology of an autoantibody to factor VIII, AVWS can be caused by a variety of mechanisms. Most of these mechanisms are due to increased clearance of VWF, either by antibodies, adsorption onto other cells, shear stress, and increased proteolytic activity, but decreased synthesis has been described as well. Unlike acquired hemophilia, which is often idiopathic, AVWS is almost always seen in association with an underlying disorder.28 The majority of AVWS has been linked to lymphoproliferative disorders, followed by other hematologic malignancies, solid malignancies, immune disorders (especially autoimmune diseases), a multiplicity of cardiac defects, infections, and certain drug use. Table 3 summarizes the different mechanisms, associated condition, and some treatment considerations.
Pathophysiologic mechanisms of AVWS, associated underlying condition, and treatment considerations
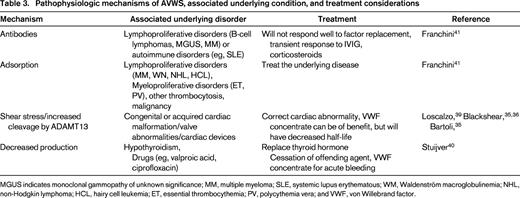
MGUS indicates monoclonal gammopathy of unknown significance; MM, multiple myeloma; SLE, systemic lupus erythematous; WM, Waldenström macroglobulinemia; NHL, non-Hodgkin lymphoma; HCL, hairy cell leukemia; ET, essential thrombocythemia; PV, polycythemia vera; and VWF, von Willebrand factor.
Laboratory diagnosis.
Depending on the pathophysiology, most cases of AVWS mimic the congenital types 1 or 2A VWD, however type 2B-like AVWS has been described.29 In conditions where accelerated clearance is implicated (immune mediated, adsorption or shear stress), there is typically loss of large multimers on electrophoresis. Although von Willebrand factor antigen (VWF:Ag), functional measurement by the Ristocetin Co factor (VWF:RCo) and collagen binding assay (VWD:CB) can be in normal range, there is a reduced activity/antigen ratio (VWF:RCo/VWF:Ag or VWF:CB/VWF:Ag). Measurement of the VWF propeptide can reflect proper synthesis and be indicative of increased VWF clearance, if the propeptide/VWF:Ag ratio is increased. Detection of actual antibodies remains challenging and not well standardized. Diagnostic laboratory evaluation for AVWS is summarized in Table 1.
Age incidence of AVWS.
Even though AVWS has been described in young children, it appears that most cases present at a more advanced age. In the largest data collection on the disorder today, the International Society of Thrombosis and Hemostasis International Registry on AVWS, the median age at presentation was 62 years. Advanced age was particularly noted in patients with underlying lymphoproliferative, cardiovascular disorders, or neoplasm (median 64, 68, and 62 years at presentation, respectively).30
Immune-mediated AVWS.
Like acquired hemophilia, AVWS can be immune mediated and is seen in association with monoclonal gammopathy of undetermined significance (MGUS), other lymphoproliferative disorders or systemic lupus erythematous. Intravenous immunoglobulin (1 g/kg/day for 2 days or 0.4 g/kg/day for 5 days) will correct laboratory abnormalities within 24-48 hours and alleviate bleeding symptoms in IgG-MGUS but not IgM-MGUS. Response can be seen for ∼21 days and periodic redosing can achieve long-term control.31 Prednisone, other immunosuppressants and rituximab have been described in the literature with varying results, it is unclear whether they work by modulating the activity of the antibodies against VWF or by controlling the potential underlying disorder.32
Association with cardiovascular disorders.
A variety of cardiovascular disorders have been associated with AVWS and can include congenital defects, acquired valve and other structural abnormalities and intracardiac devices. Approximately 20% of adults with congenital heart disease have AVWS.33 Affected individuals have a decrease of high molecular weight VWF multimers as a result of increased shear stress and subsequent mechanical demolition and easier proteolysis by ADAMTS13.34 Usually that does not result in abnormal VWF:Ag, VWF:RCo, or VWF:CB levels but the VWF:RCo/Ag and VWF:CB/Ag ratio is often reduced. The question is whether these changes in multimers result in phenotypic bleeding. A study evaluating patients with hypertrophic cardiomyopathy showed that VWF levels were not predictive of bleeding, but older age, female gender, and higher peak outflow velocity measurements were.35 A smaller study in mitral regurgitation also showed more bleeding with older age. This study actually showed higher VWF:Ag or VWF:Act levels in the bleeding patients but showed a significantly lower VWF:Act/VWF:Ag ratio and that this ratio decreased with MR severity.36 A study of 41 consecutive patients needing valve replacement for aortic stenosis reported that 17% had mild bleeding symptoms leading up to it. There was loss of large VWF multimers in 80.5% and a decreased VWF:CB/VWF:Ag ratio. Bleeding was more likely to occur in patients with higher transvalvular gradients and was correlated with marked loss of large multimers. The benefit of VWF containing concentrates is limited for the treatment of bleeding as the VWF proteins in the concentrate are exposed to the same destructive forces as the native proteins. It is reasonable to administer VWF concentrate immediately pre-operatively to prevent bleeding, but increased clearance and frequent redosing has to be anticipated. The abnormality causing the VWF destruction is corrected within the first day of surgery, tends to last for up to 6 months postoperatively,37 but can recur with recurring cardiac abnormality.
Heyde's syndrome.
A syndrome of gastrointestinal bleeding in association with calcific aortic stenosis in elderly people was first reported in 1958 by E. C. Heyde.38 Loss of large VWF multimers has been described since and is thought to be due to the above noted shear force and cleavage of VWF by ADAMTS13. The associated angiodysplasia is not fully understood but may present a combination between normal vascular aging and an impairment of platelets to maintain vascular endothelium. Bleeding symptoms usually resolve after valve replacement.39
Association with hypothyroidism.
A recent prospective study examined 90 consecutive adult patients with newly diagnosed overt hypothyroidism and found that 33% had VWF:Ag and/or VWF:RCo levels of ≤50%. Most of them only had mild decreases with levels between 30% and 50% and none of them were severe (levels <10%). This appeared to be a quantitative defect where VWF levels correlated with free T4 levels and mucocutaneous bleeding symptoms were increased in patients with lower VWF levels. Almost all VWF levels rose in response to thyroid replacement therapy.40
General treatment considerations.
The treatment of AVWS and associated bleeding is ultimately to address the underlying disorder. It is crucial to consider the causal pathophysiology when treating acute bleeding, as it will determine the most appropriate approach. Desmopressin (DDAVP) and VWF/FVIII replacement, for example, have been shown to have limited hemostatic efficacy when VWF is impaired due to destruction. On the other hand, replacement therapy can be very useful if AVWS is caused by underproduction. In immune mediated AVWS, immunoglobulin infusions may have a rapid effect. In severe cases the use of recombinant activated factor VII has been reported.41
Conclusions
Older patients, especially those on antithrombotic therapy, should routinely be screened for new bleeding symptoms. The physiology and environment of aged patient is subject to change over time, leading to supratherapeutic levels of these medications. In addition, the aged population is at increased risk for coagulation disorders such as acquired hemophilia or acquired von Willebrand syndrome, which can be challenging to manage, particularly in a prothrombotic environment. Astute awareness and early detection will likely decrease morbidity and mortality in the elderly with acquired bleeding disorders.
Correspondence
Rebecca Kruse-Jarres, Washington Center for Bleeding Disorders at Bloodworks Northwest, University of Washington, 921 Terry Ave, Seattle, WA 98104; Phone: 206-689-6507; Fax: 206-689-8341; e-mail: rebeccakr@bloodworksNW.org.
