
Abstract
Atypical chronic myeloid leukemia (aCML) and chronic neutrophilic leukemia (CNL) are rare myeloid neoplasms defined largely by morphologic criteria. The discovery of CSF3R mutations in aCML and CNL have prompted a more comprehensive genetic profiling of these disorders. These studies have revealed aCML to be a genetically more heterogeneous disease than CNL, however, several groups have reported that SETBP1 and ASXL1 mutations occur at a high frequency and carry prognostic value in both diseases. We also report a novel finding—our study reveals a high frequency of U2AF1 mutations at codon Q157 associated with CSF3R mutant myeloid neoplasms. Collectively, these findings will refine the WHO diagnostic criteria of aCML and CNL and help us understand the genetic lesions and dysregulated signaling pathways contributing to disease development. Novel therapies that emerge from these genetic findings will need to be investigated in the setting of a clinical trial to determine the safety and efficacy of targeting various oncogenic drivers, such as JAK1/2 inhibition in CSF3R-T618I–positive aCML and CNL. In summary, recent advances in the genetic characterization of CNL and aCML are instrumental toward the development of new lines of therapy for these rare leukemias that lack an established standard of care and are historically associated with a poor prognosis.
Learning Objectives
Compare and contrast the WHO diagnostic criteria for atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Describe the clinical features and therapeutic challenges in atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Describe the most common molecular drivers in atypical chronic myeloid leukemia and chronic neutrophilic leukemia and the signaling pathways leading to leukocytosis
Atypical chronic myeloid leukemia (aCML) and chronic neutrophilic leukemia (CNL) are rare myeloid leukemias typically associated with a poor prognosis and without established standards of care. Therapies used in other myeloproliferative neoplasms have not been uniformly successful in controlling leukocytosis, reducing spleen volume, improving symptoms, or inducing remission. The recent discovery of mutations in colony-stimulating factor 3 receptor (CSF3R) at a high frequency in CNL and at a lower frequency in aCML has prompted a more comprehensive survey of the molecular landscape of these leukemias.1,2 Several groups have suggested substantial revisions to the WHO diagnostic criteria based on these findings. It has become increasingly clear that the genetic drivers and pathologic features are heterogeneous in aCML but less so in CNL. Collectively, these new insights will improve our understanding of disease pathogenesis and help tailor our efforts toward effective therapies.
Diagnosing atypical chronic myeloid leukemia
The blood smear of patients with CML and aCML at a first glance may look indistinguishable with both diseases displaying prominent immature granulocytosis (promyelocytes, myelocytes, and metamyelocytes; Figure 1). Patients with this blood smear but without the Philadelphia chromosome or BCR-ABL translocation are diagnosed as having “atypical” chronic myeloid leukemia. This term is not used to describe CML patients with variant BCR-ABL transcripts, such as the p230 BCR-ABL. The WHO has defined specific morphologic criteria in the blood and marrow for the diagnosis of aCML, in addition to the absence of BCR-ABL transcripts and PDGFRA, PDGFRB, and FGFR1 rearrangements.3 The latter group of rearrangements is associated with eosinophilic leukemias.

Typical smear of blood and marrow aspirate in CML, aCML, and CNL. (A) CML peripheral blood smear. Morphologically unremarkable neutrophils with frequent immature granulocytes, including a disproportionally high percentage of myelocytes. Basophilia, thrombocytosis, and eosinophilia are also common. (B) Aspirate smear. Myeloid hyperplasia without overt granulocytic dysplasia, frequent myelocytes (so-called “myelocyte bulge”) and marrow eosinophilia. Note the micromegakaryocyte or “dwarf megakaryocyte” in the lower left-corner. (C) aCML peripheral blood smear. Frequent dysplastic neutrophils; note the dysplastic morphologic features including hypogranularity and hypolobation (pseudo-Pelger–Huët anomaly, inset). (D) aCML aspirate smear. Myeloid hyperplasia with prominent dysplasia and without basophilia/eosinophilia. (E) CNL peripheral blood smear. Neutrophilia without dysplastic forms or immature granulocytes. (F) CNL Aspirate smear. Myeloid hyperplasia without dysplastic morphologic features or basophilia/eosinophilia. Images obtained at 63×, courtesy of Dr Philipp W. Raess.
Typical smear of blood and marrow aspirate in CML, aCML, and CNL. (A) CML peripheral blood smear. Morphologically unremarkable neutrophils with frequent immature granulocytes, including a disproportionally high percentage of myelocytes. Basophilia, thrombocytosis, and eosinophilia are also common. (B) Aspirate smear. Myeloid hyperplasia without overt granulocytic dysplasia, frequent myelocytes (so-called “myelocyte bulge”) and marrow eosinophilia. Note the micromegakaryocyte or “dwarf megakaryocyte” in the lower left-corner. (C) aCML peripheral blood smear. Frequent dysplastic neutrophils; note the dysplastic morphologic features including hypogranularity and hypolobation (pseudo-Pelger–Huët anomaly, inset). (D) aCML aspirate smear. Myeloid hyperplasia with prominent dysplasia and without basophilia/eosinophilia. (E) CNL peripheral blood smear. Neutrophilia without dysplastic forms or immature granulocytes. (F) CNL Aspirate smear. Myeloid hyperplasia without dysplastic morphologic features or basophilia/eosinophilia. Images obtained at 63×, courtesy of Dr Philipp W. Raess.
Another defining feature in aCML, not present in CML or CNL, is prominent granulocytic dysplasia. Evidence for dysplasia in the neutrophils includes pseudo-Pelger–Huët forms or other abnormal segmentation of nuclei, abnormal chromatin clumping, or reduced or abnormal cytoplasmic granularity. Although “prominent” dysgranulopoiesis is considered a “major” feature according to the WHO diagnostic criteria, this criterion is not defined specifically in quantitative terms.3 One might conservatively assume that dysgranulopoiesis should occur in >10% of the cells, similarly to the 2008 WHO criteria for the bone marrow findings in myelodysplastic syndromes.
In Table 1, we compare and contrast the blood and marrow findings in CML, aCML, and CNL as defined by the 2008 WHO diagnostic criteria.3 Because there are no defining mutations in aCML, the diagnosis is strictly based on morphologic evaluation of the blood and marrow and may pose diagnostic challenges in clinical practice. For example, some patients may not meet the criteria for having aCML based on the percentage of immature granulocytes in the blood, which can fluctuate widely due to a variety of reasons and may not reflect a true hallmark of biologically distinct disease. With more comprehensive molecular profiling, the diagnostic criteria will be refined in the future to also include genetic features resulting in decreased reliance on absolute cutoffs.
Summary of 2008 WHO diagnostic criteria

nd, not defined.
* 90%-95% have the t(9;22)(q34;q11.2) there are rare cases of variant translocations or cryptic translocations; absolute basophilia is invariably present and eosinophilia is common. Small, hypolobated megakaryocytes (“dwarf megakaryocytes”) are usually present.
† In the absence of a clonal cytogenetic or molecular marker, rule out reactive neutrophilia and an underlying tumor.
‡ Promyelocytes, myelocytes, and metamyelocytes.
It is important to distinguish aCML from immature granulocytosis induced by other neoplastic disorders (eg, CML, cellular phase of primary myelofibrosis, or marrow infiltration by carcinomas) and non-neoplastic conditions (inflammatory states, infectious causes, and toxic marrow damage). These conditions may lead to a leukemoid reaction or a leukoerythroblastosis blood smear. The clinical circumstances, such as onset of abnormalities, duration of leukocytosis, presence of splenomegaly, and other coexisting acute or chronic medical issues, may provide initial clues but ultimately a bone marrow evaluation and cytogenetic analysis in suspected cases are required. Incorporation of molecular studies may be very helpful to distinguish between benign versus malignant causes of leukocytosis.
aCML is commonly associated with cytogenetic abnormalities in as many as 80% of cases but there are no specific recurring cytogenetic findings that define aCML. The most frequent abnormalities are trisomy 8 and deletion 20q, but a variety of translocation and aneuploidy abnormalities have been reported in case reports and small case series. The frequency of CSF3R-T618I mutations in aCML is anywhere from 0% to 40%.1,2,4,5 SETBP1 mutations co-occur at a high frequency.5,6 The stringency with which one applies the 2008 WHO diagnostic criteria may represent one factor in the wide spectrum of frequencies reported.
Diagnosing chronic neutrophilic leukemia
The characteristic feature of CNL is neutrophilia in the absence of prominent immature granulocytosis and granulocytic dysplasia. The WHO diagnostic criteria for CNL are listed in Table 1. Mutations in CSF3R were recently found at a high frequency in CNL. Several additional datasets indicate that the prevalence of CSF3R mutations in CNL may be as high as 100% if the WHO criteria are strictly applied.2,7 Several groups have suggested a revision of the WHO criteria to incorporate CSF3R mutations as part of the diagnostic criteria of CNL. By incorporating CSF3R mutation as a major criterion for diagnosing CNL, some of the more arbitrary criteria, such as WBC >25 × 10e9/L, neutrophils plus bands >80% of total WBCs, and the less-defined criteria of hepatosplenomegaly may no longer be necessary.
There are many causes of neutrophilia in isolation. Because dysplasia is not a defining morphologic feature in CNL and cytogenetics are normal in approximately 90% of cases, these findings do not help distinguish CNL from nonmalignant causes or neutrophilia caused by a G-CSF-secreting tumor. All patients should be clinically evaluated for chronic infectious or inflammatory conditions. In the past, clonal disease in the neutrophils was demonstrated by the presence of nonrandom inactivation of the X-linked HUMARA gene. However, this test is not routinely available in the clinical setting and there are nonmalignant causes of clonal hematopoiesis. Measurement of G-CSF and GM-CSF produced by solid tumors is also not routinely available. Having a documented CSF3R mutation may help rule-in a diagnosis of CNL, but the absence of a CSF3R mutation does not categorically rule-out CNL.
There is a remarkable association between CNL and plasma cell dyscrasias based on numerous case reports and small case series in the literature. Some of these cases were felt to be secondary to the myeloma, whereby the neoplastic plasma cells produce abnormal cytokines leading to neutrophilia or exert its effects indirectly through other cells. Now that CSF3R and other CNL-associated mutations have been identified, it will be interesting to determine whether the neutrophilia is truly reactive or representative of a biphenotypic neoplasm.
Overlapping clinical characteristics and prognosis in aCML and CNL
Leukocytosis is a common finding in both aCML and CNL. Leukocyte alkaline phosphatase may be normal or elevated and is not uniformly helpful, especially in the current era of molecular testing. Hyperleukocytosis can be very prominent raising the concern for leukostasis. However, like CML, many patients tolerate very high leukocyte counts without having any symptoms or direct evidence of end organ damage or perfusion defects. The insidious onset may preclude an accurate assessment of when the disease started and the natural history of disease. Sometimes the discovery of leukocytosis may be incidental (on a routine annual physical examination) or may be part of a work-up for signs or symptoms related to disease, such as weight loss, fatigue, anemia, night sweats, pruritus, easy bruising or bleeding, bone pain, or abdominal pain. The use of a symptom assessment tool (eg, MPN-SAF)8 may help clinicians address symptom burden in patients and provide standardized prospective data on whether therapy is effective (on or off clinical trial). Varying degrees of anemia and thrombocytopenia are present. Many of these clinical features overlap with the cellular phase of primary myelofibrosis and may be particularly challenging morphologically to differentiate from aCML and MPN not otherwise specified. Primary myelofibrosis is associated with JAK2-V617F or CALR mutations in ∼90% of cases. Morphologic features that are more prominent in primary myelofibrosis than in aCML and CNL include megakaryocytic hyperplasia with atypia, reticulin and/or collagen fibrosis, and a leukoerythroblastic blood smear.
Bleeding diatheses are common in aCML and CNL. Patients experiencing excessive bruising and bleeding out of proportion to their platelet count should be worked up for acquired von Willebrand's disease and other acquired coagulation factor or platelet function defects. Neutrophilic infiltration and destruction of vessels has been reported in a patient with severe bleeding issues.9 Functional neutrophil abnormalities have also been described.
Splenomegaly is a common finding in both aCML and CNL and may range from mild to massive splenomegaly. Symptoms associated with splenomegaly may include weight loss, early satiety, and abdominal fullness or pain. Hepatomegaly may or may not be clinically appreciated by physical exam. In CNL, tissue infiltration by neutrophils is felt to be the predominant process of organomegaly. Skin findings may include leukemia cutis, Sweet's syndrome, and pyoderma gangrenosum.
In all patients, the standard age-appropriate cancer screening and risk or symptom-adapted screening should be completed based on the rare occurrence of G-CSF- and GM-CSF-secreting tumors.
Though no distinct phases of disease have been assigned to aCML and CNL, these diseases frequently demonstrates an accelerated phase associated with increased WBC, progressive hepatosplenomegaly, worsening cytopenias, and increased blasts. This may or may not be associated with new cytogenetic or molecular markers (models presented in Figure 2). In the largest series of WHO-defined aCML, the median survival was 25 months in 55 patients.10 Shorter survival was associated with age >65 years old, female sex, WBC >50 × 10e9/L, and the presence of immature granulocytosis in a multivariate analysis.10 Immature granulocytosis was not precisely stated in this study, since by WHO-defined criteria, all cases should demonstrate immature granulocytosis. Elliott et al evaluated 12 patients with CNL and comprehensively reviewed the literature and reported that the median survival was 23.5 months in 40 cases of WHO-defined CNL.11 Common causes of death include intracerebral hemorrhage, progressive bone marrow failure and its complications, blastic transformation, and progressive multi-organ failure.

Various models of disease evolution in CSF3R-T618I–positive myeloid neoplasms. We compare and contrast to CML (Model 1). Nearly all patients with WHO-defined CNL harbor a CSF3R-T618I mutation (Model 2) according to some datasets. These patients may undergo similar pathologic/disease phases as CML and the transition points are associated with additional genetic changes. It is unclear whether CSF3R-T618I–positive aCML is a distinct entity (Model 3) or a disease evolution from CNL (Model 4). A number of oncogenic drivers are possible in aCML (Table 2). Future studies will help clarify the leading model in aCML. HSC indicates hematopoietic stem cell. The yellow lightning symbol depicts the acquisition of somatic mutation(s).
Various models of disease evolution in CSF3R-T618I–positive myeloid neoplasms. We compare and contrast to CML (Model 1). Nearly all patients with WHO-defined CNL harbor a CSF3R-T618I mutation (Model 2) according to some datasets. These patients may undergo similar pathologic/disease phases as CML and the transition points are associated with additional genetic changes. It is unclear whether CSF3R-T618I–positive aCML is a distinct entity (Model 3) or a disease evolution from CNL (Model 4). A number of oncogenic drivers are possible in aCML (Table 2). Future studies will help clarify the leading model in aCML. HSC indicates hematopoietic stem cell. The yellow lightning symbol depicts the acquisition of somatic mutation(s).
Advances in defining molecular drivers and disease modifying mutations in aCML and CNL
The most common mutation in CSF3R is an activating point mutation in the membrane proximal region (CSF3R-T618I; also known as T595I if the signal peptide is excluded from the numbering), which activates JAK/STAT signaling through increased receptor dimerization resulting in ligand independence.1,12 Other much less common activating point mutations include T615A and T640N. Another class of mutations (nonsense and frameshift mutations) results in truncation of the cytoplasmic tail of CSF3R.7 Truncating mutations have been previously reported in ∼30% of patients with severe congenital neutropenia (SCN) undergoing long-term G-CSF therapy. SCN patients often harbor these truncating mutations for years without malignant transformation. However, these patients are at increased risk for development of AML, and the occurrence of AML has been shown to coincide with acquisition of secondary membrane proximal point mutations in CSF3R. Compound mutations of both truncating and membrane proximal mutations have also been observed in aCML and CNL cases.1
In vivo studies confirm that the CSF3R-T618I mutation is an oncogenic driver.13 In a bone marrow transplant model, all mice developed a lethal neutrophilic leukemia with extensive liver and spleen infiltration of neutrophils. Treatment of mice with a JAK1/2 inhibitor (ruxolitinib) reduced disease burden and improved survival of mice. In unpublished work by the same authors, truncating mutations of CSF3R did not produce disease in mice and is entirely consistent with the SCN data. Interestingly, patients with autosomal dominant hereditary neutrophilia due to a germline CSF3R-T640N mutation (also known as T617N) do not uniformly develop hematologic malignancies, suggesting that this mutation by itself does not produce malignant transformation without other disease modifying or cooperating mutations.
Although CSF3R-T618I almost uniformly defines WHO-defined CNL, a number of oncogenic drivers seen in aCML are neither specific to aCML nor present in the majority of aCML cases. Mutations have been reported in CALR, CBL, CSF3R, ETNK1, JAK2, KIT, MPL, and RAS (see Table 2 for summary and references). The rare occurrence of aCML limits some studies to small case series and the variability in applying the WHO-defined criteria account for some of the wide range of mutation frequencies for some of these genes. We include in Table 2 the disease produced by each oncogenic driver in animal models and the candidate inhibitors for each oncogenic pathway.
Oncogenic driver mutations reported in aCML and their functional consequences

* Unpublished data from Jeffrey Tyner, May 2015.
Mutations in SETBP1 are very frequent in aCML (0%-33%)2,5,6 and in CNL (14%-75%).2,5,7,14 SETBP1 mutations also occur in CMML. SETBP1 mutations usually occur between codons 858 and 871 within the SKI homologous region.6 These same mutations occur in Schinzel–Giedion syndrome, a congenital condition due to de novo somatic mutations in SETBP1 leading to severe abnormalities in organ development and increased risk for neuroepithelial tumors. The most common mutations, D868N, G870S, and I871T, disrupt ubiquitination and reduce turnover of SETBP1. The stabilization of SETBP1 protects SET from protease degradation, which in turn inhibits PP2A activity and induces proliferation signals. In several case series, SETBP1 mutations are thought to be associated with inferior survival.
ASXL1, which regulates histone modification, is also mutated at a high frequency; in one study, 66% (38 of 58) of aCML, 57% (8 of 14) of CNL, and 45% (66 of 146) of CMML.5 The high frequency of ASXL1 mutations in CNL was confirmed in another report and has been shown to confer negative prognostic impact in CNL and in other myeloid neoplasms.15,16 ASXL1 mutations are commonly frameshift and nonsense mutations in exon 12 leading to a truncated gene product. The mutation present in 50% of cases is a duplication of a guanine nucleotide at position 1934. An ASXL1 knockout animal model displayed mild abnormalities in hematopoiesis.17 Because myeloid malignancies did not develop in this model, this suggests that ASXL1 mutation is a disease-modifying mutation.
We report on 10 cases of CSF3R-T618I–positive myeloid neoplasms at our institution and list the various mutations found in addition to the CSF3R-T618I mutation (Table 3). We obtained IRB approval for this limited review. Mutations in SETBP1 and ASXL1 occurred in ∼30% of cases for each gene. Interestingly, we identified frequent mutations in U2AF1 (4 of 10 cases of chronic myeloid neoplasms, not defined further in this study). There was no mention of mutations in U2AF1 in the datasets that focused on aCML and CNL but the targeted sequencing panel did not appear to include U2AF1.2,5 In a study that performed whole exome sequencing of 8 aCML samples, U2AF1 mutations were not reported.6 U2AF1 mutations have been reported in primary myelofibrosis (15%), myelodysplasia without ring sideroblasts (12%), and CMML (8%).18 U2AF1 is an accessory factor in the U2 snRNP component of the spliceosome. Mutations in U2AF1 occur in the first (eg, codon S34) or second (eg, codon Q157) zinc finger domains and have been shown to alter splicing of many important genes in myeloid disorders.19 The high frequency of U2AF1 mutations at codon Q157 in CSF3R-T618I–positive myeloid neoplasms is a novel finding but the prognostic significance remains unknown at this time.
Mutations co-occurring with CSF3R mutations in myeloid neoplasms
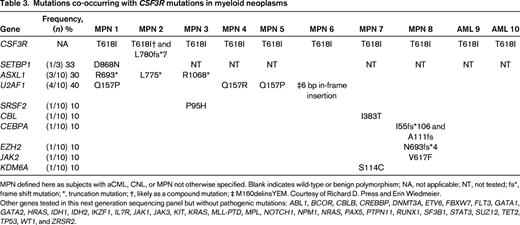
MPN defined here as subjects with aCML, CNL, or MPN not otherwise specified. Blank indicates wild-type or benign polymorphism; NA, not applicable; NT, not tested; fs*, frame shift mutation; *, truncation mutation; †, likely as a compound mutation; ‡ M160delinsYEM. Courtesy of Richard D. Press and Erin Wiedmeier.
Other genes tested in this next generation sequencing panel but without pathogenic mutations: ABL1, BCOR, CBLB, CREBBP, DNMT3A, ETV6, FBXW7, FLT3, GATA1, GATA2, HRAS, IDH1, IDH2, IKZF1, IL7R, JAK1, JAK3, KIT, KRAS, MLL-PTD, MPL, NOTCH1, NPM1, NRAS, PAX5, PTPN11, RUNX1, SF3B1, STAT3, SUZ12, TET2, TP53, WT1, and ZRSR2.
SRSF2 and TET2 are also frequently mutated in aCML and CNL but will not be discussed here due to space limitations.5
Clinical management and new directions in aCML and CNL
There are no standard of care guidelines established for aCML and CNL. Therapies effective in other MPNs have been met with widely varying success in controlling leukocytosis, symptoms, and splenomegaly. There is very limited data on therapeutic responses and many of the experience is anecdotal.14,20 Some of these measures include hydroxyurea, JAK1/2 inhibitor (ruxolitinib), imatinib or second-generation inhibitors, interferon-alpha, hypomethylating agents, thalidomide or lenalidomide, histone deacetylase inhibitors, and chemotherapy agents (eg, 6-thioguanine, cytarabine, cladribine, busulfan, and standard AML “7 + 3” induction chemotherapy). Molecular profiling may ultimately allow for implementation of molecularly targeted therapy (Table 2). However, at the present time this is not recommended routinely outside of a clinical trial. Our group is conducting a clinical trial to study the safety and efficacy of ruxolitinib in aCML and CNL patients with or without CSF3R mutations (NCT02092324). Additionally, we aim to define the genetic determinants of clinical responses to ruxolitinib.
Of particular interest are the effects of biologic therapies in aCML and CNL, for example, interferon-alpha, hypomethylating agents, and TNF-alpha inhibitors. Interferon-alpha is known to modulate the immune system, stromal cells, and neoplastic cells and can induce hematologic and molecular remissions in Philadelphia-negative MPN patients. The pegylated formulations of interferon-alpha have an improved toxicity profile and may allow one to extend this treatment option to more patients. Azacitidine is a hypomethylating agent known to improve overall survival of high-risk MDS patients compared with the conventional care arm by a median of 9.5 months.21 Testing the safety and efficacy of azacitidine is certainly a concept to consider especially in aCML where there are overlapping morphologic, molecular, and cytogenetic features with MDS. Anti-TNF-alpha therapies may also be worth investigating because preclinical studies have shown that TNF-alpha induces neoplastic clonal outgrowth and inhibits normal hematopoiesis.22,23 Another intriguing concept is the role of inhibitors of apoptosis in neutrophil accumulation in CNL.24 We propose that future clinical trial studies should consider molecular profiling of aCML and CNL followed by combining an inhibitor of an oncogenic pathway with or without biologic therapies that might have clinical benefits beyond palliative care. Considering that these diseases are so rare, it will be challenging to determine whether intervention with any of these agents alter the natural history and provide survival benefit. In the absence of a clinical trial, we suggest using hydroxyurea to control leukocytosis and referring eligible patients for transplant evaluation. We refrain from providing specific advice on which second line agent should be used given the lack of data on safety and efficacy of these agents in the setting of a clinical trial.
Myeloablative or reduced intensity conditioning regimens followed by allogeneic stem cell transplant is the only curative option for a minority of patients. In small series combining a variety of Philadelphia-negative CML and MDS/MPN disorders, the long-term survival at 5 years is ∼40%-50%.25,26 In 9 patients with aCML, the outcome was favorable for nearly all of the patients with a median follow-up of 55 months.27 Persistence of molecular markers after allogeneic stem transplant is associated with increased risk of disease relapse28 and is a major cause of treatment failure in reduced intensity conditioning allogeneic stem cell transplant. In all case series evaluating survival in aCML and CNL, there is a subgroup of patients that have a more indolent clinical course with occasional survival of over 5 years. Hopefully, with better genetic characterization of these leukemias, we will be able to define this subgroup more precisely and reserve allogeneic stem cell transplant for disease progression. Patients with SETBP1 and/or ASXL1 mutations appear to have a worse prognosis, and in such cases, in eligible patients, allogeneic stem cell transplant should be considered as upfront disease management.
Splenectomy has a high perioperative mortality rate in MPN patients in general and has been associated with accelerated neutrophilia anecdotally. Splenectomy is not recommended for management of disease. Splenic irradiation may provide temporary palliative benefit but may worsen cytopenias and/or increase risk for clonal evolution and secondary malignancies. Reserving this treatment modality primarily for palliative reasons in patients not expected to live beyond 6 months might be appropriate.
In patients who have documented myeloma in the setting of a confirmed or not confirmed myeloid neoplasm (CNL) with evidence of end-organ damage, standard myeloma therapies may be given with additional clinical considerations if there are preexisting cytopenias and bleeding issues.
In patients whose disease course has changed based on laboratory findings, progressive hepatosplenomegaly, increased symptom burden, or other clinical findings, a bone marrow evaluation along with cytogenetic and molecular studies is recommended. There have been case reports of CNL occurring in patients with a prior hematologic malignancy, such as polycythemia vera and myelodysplasia.
Summary
The WHO criteria for diagnosing aCML and CNL will likely be updated in the next iteration to include new genetic features of aCML and CNL. Applying the current WHO-defined criteria has posed practical challenges as some criteria are subject to interpretation. We anticipate the new version of WHO diagnostic criteria for CNL will be less reliant on arbitrary cutoffs of morphologic and laboratory parameters with greater inclusion of recent genetic findings. We suggest the diagnosis of CNL can be made when all five major criteria are met, including: (1) presence of a disease-associated CSF3R mutation, (2) neutrophilia in the absence of prominent dysplasia (<10% in blood and marrow), (3) absence of immature granulocytosis (<10% in blood), (4) blasts <5% in blood and marrow, and (5) no mutations in BCR-ABL, PDGFRA, PDGFRB, or FGFR1. For the minority of cases with wild-type CSF3R, we suggest that all of the other major criteria must be met, in addition to the presence of another candidate driver, such as JAK2-V617F. In contrast, the diagnostic criteria for aCML will likely continue to emphasize morphologic criteria in the blood and marrow because there is not a single most common oncogenic driver (Table 2). We suggest that the presence of one of these oncogenic drivers or other rare drivers is listed as a major criterion for diagnosing aCML. For both aCML and CNL, we suggest mentioning that SETBP1, ASXL1, and U2AF1 mutations are frequent disease-modifying mutations in these disorders. Mutant SETBP1 and ASXL1 have prognostic value, whereas mutant U2AF1 has not been studied yet in aCML and CNL but should be investigated further.
In Figure 2, we summarize possible models of disease development for aCML and CNL as they relate to well-developed models of CML. Although CML is known to occur as a result of BCR-ABL acquisition within an early hematopoietic stem/progenitor cell with additional genetic lesions contributing to the accelerated phase and blast crisis phase (Model 1), the evolution of aCML and CNL, as well as the sequence of acquisition of genetic events is less clear. Based on collective anecdotal experience and the literature, it seems likely that multiple scenarios occur. One possibility is that CSF3R mutations are acquired similarly to BCR-ABL at an early stem/progenitor stage and disease progresses through similar phases, with acquisition of additional genetic abnormalities (Figure 2, Models 2 and 3). It also seems possible that CSF3R-T618I–positive aCML is part of the disease evolution of CNL as shown in Model 4. In other words, CSF3R-T618I–positive aCML may represent an accelerated phase of CNL. Another scenario is that alternative genetic events (such as SETBP1 and ASXL1) occur first, creating a damaged hematopoietic stem cell and/or bone marrow microenvironment that increases the risk for acquiring CSF3R or other MPN-associated mutations (eg, CBL or RAS) in a lineage-restricted progenitor. This could explain why different clinical and cellular phenotypes are possible in aCML, CMML, and MDS/MPN despite all of these diseases sharing a high frequency of mutations in SETBP1 and ASXL1.
Currently available data preclude a conclusion that would definitively favor one of these models, but ongoing studies will illuminate the clonal architecture and evolution of disease driven by the acquisition of genetic events. In the era of cancer genetics, there should be substantial progress in the coming years with tailoring therapies toward the cancer genomes of aCML and CNL. The order of mutational acquisition and the number and type of mutations may reveal an opportunity for early intervention with improved clinical outcomes. Additional clinical trials will be needed to risk-stratify patients and to investigate the safety and efficacy of disease-modifying therapies.
Acknowledgments
The authors thank Drs Julia Maxson, Jason Gotlib, and Daniel Pollyea for their contribution to this research; and Drs Philipp W. Raess, Richard D. Press, and Erin Wiedmeier for the data contributed. This work was supported by the Oregon Health & Science University Knight Cancer Institute and the National Institutes of Health/National Heart, Lung, and Blood Institute (1K08HL111280 to K.T.D.), grants from The Leukemia & Lymphoma Society, National Institutes of Health/National Cancer Institute (1R01CA183947 and 5R00CA151457 to J.W.T.), V Foundation for Cancer Research (J.W.T.), and Gabrielle's Foundation for Cancer Research (J.W.T.). We sincerely apologize that we could not cite every reference in support of the content of this article due to space limitations.
Correspondence
Kim-Hien Dao, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Mailcode UHN73C, Portland, OR 97239; Phone: 503-494-7894; Fax: 503-494-3688; e-mail: daok@ohsu.edu.
