
Abstract
A 68-year-old male with history of hypertension and arthritis presented with bruising and increasing fatigue over several months. He was found to be thrombocytopenic (platelets 30), WCB 2.0 K/mm3, Hg 11.6 g/dL, ANC 870, and 1% circulating blasts. Bone marrow biopsy revealed 40%-50% cellular with multilineage dysplasia and 10% blasts. Cytogenetic genetic studies showed trisomy 2, and translocation (3;21). FISH studies for 5q, 7p, 8, 17p, and 20q abnormalities were negative. Molecular diagnostics were sent to a commercial laboratory to aid in prognostication. These studies showed mutations in TET2 (exons 1- 9 tested) and PHF6 (exons 1-9 tested). No abnormalities in other epigenetic regulators (DNMT3A, ASXL1), RNA splicing (SF3B1, SRSF2, URAF1, ZRSR2), transcription factors (RUNX1 or ETV6), or signaling (CBL, NRAS, KIT, JAK2, MPL) were detected. He was referred for consultation regarding initial treatment. In this elderly patient with preserved organ function and good performance status who is being considered for reduced intensity conditioned allogeneic hematopoietic cell transplant, what should the initial treatment be and can we use the molecular diagnostic results to guide therapy?
Learning Objective
To review the role somatic mutational testing has at diagnosis for patients with MDS to predict response to standard therapies
Discussion
The patient in this vignette presented with de novo myelodysplastic syndrome (MDS), RAEB-2 with R-IPSS of high risk.1 This portends a median overall survival of 1.6 years and time to 25% of patients developing acute myeloid leukaemia (AML) of 1.4 years.1 Although survival results may be improved by active treatment,2,3 there is little hope for longer-term disease-free survival without hematopoietic cell transplantation. Retrospective reports suggest that bone marrow blast percentage <5%-10% at the time of transplant may improve relapse-free survival after transplant.4-10
Despite current clinical prognostication systems in MDS helping to predict survival and risk for evolution to acute leukemia; predicting response to treatment has been based on a number of clinical factors including: baseline bone marrow fibrosis, increased bone marrow blasts >15%, cytogenetics, prior therapy, transfusion dependence, and doubling of platelets with therapy.3,11-14 The understanding of MDS at the genomic level has enlightened our understanding of cellular pathways recurrently mutated in MDS including RNA splicing factors, DNA methylation, transcription regulation, chromatin and histone modification, DNA repair and tumor suppressors, signal transduction, and cohesion complex abnormalities.15-22 More than 80% of patients with MDS harbor a recurrent somatic mutation and these mutations have clear prognostic significance whether in dominant clones or subclones.15,20,23 The challenges of interpreting molecular genetic data for individual patients has been reviewed previously.24 These challenges include: (1) the polyclonal nature of MDS means that multiple clones can harbor different mutational profiles and those that might predict response may not predict response if only found in a small subclone. (2) Genes can be mutated in distinct patterns and not all mutations in a gene may be equal. (3) Mutations co-occur in MDS and the interactions of mutations in common or different cellular pathways and the interaction of these mutations with clinical and cytogenetic abnormalities is poorly understood.
Despite these current knowledge gaps and these limitations, evaluating for molecular predictors of response to therapy remains an attractive and active area of research in MDS through retrospective reviews of patients previously treated and prospective observations in ongoing trials. The standard treatment for MDS remains hypomethylating agents (HMAs), azacitidine and decitabine, which inhibit DNA methyl transferase. This review will summarize our current understanding and level of evidence for selecting therapies based on the molecular genetics of MDS.
To evaluate the impact of somatic mutations in MDS on response to therapy, a PubMed search was performed. The following search criteria were used: [“Myelodysplastic Syndromes” (Mesh) AND “Mutation”(Mesh) AND (“last 5 years” (PDat) AND Humans (Mesh) AND English(lang)]. This initially returned 777 hits, 127 reviews, and 62 clinical trials. The search was limited to publication dates within 5 years, Humans, and English. Phase I trials, studies specifically in del5q MDS, MDS/MPN overlap or CMML, pharmacokinetic studies, case series, and non-MDS investigations were excluded. There are no randomized controlled clinical trials evaluating the response to therapy based on somatic mutations in MDS. Table 1 lists the 25 studies included in this review and their characteristics.
Somatal mutations in MDS and response to therapy
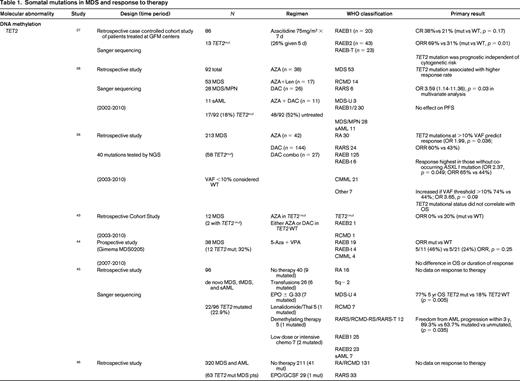
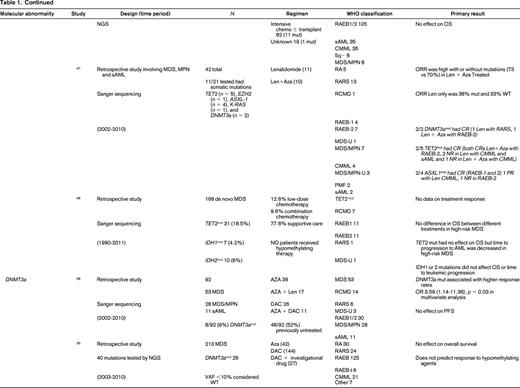

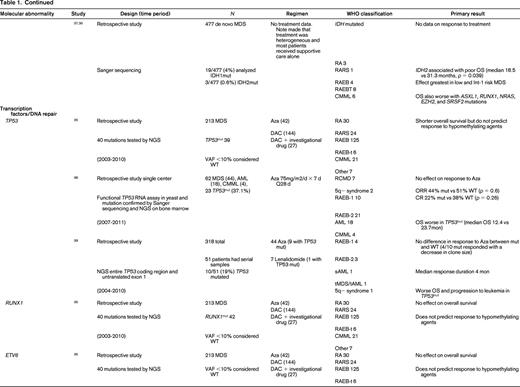
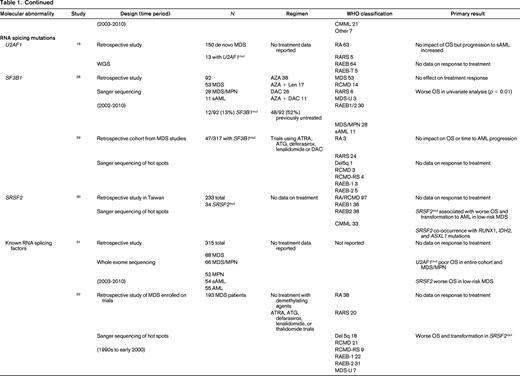
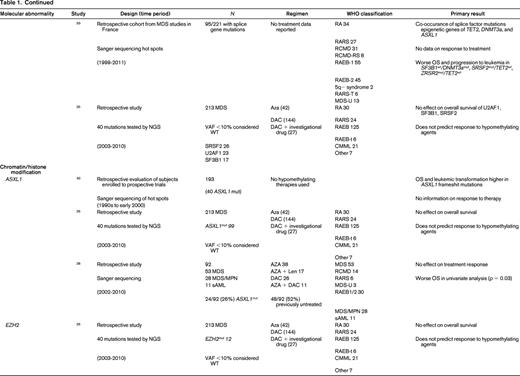
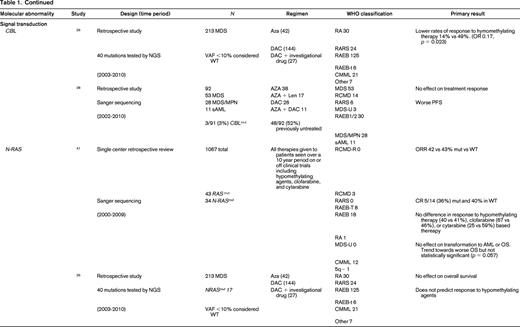

AZA indicates 5-azacitidine; DAC, decitabine; Len, lenalidomide; MDS, myelodysplastic syndrome; ATRA, all-trans retinoic acid; ATG, antithymocyte globulin; VPA, valproic acid; EPO, erythropoietin; G, neupogen/G-CSF; MPN, myeloproliferative neoplasm; NGS, next generation sequencing; RA, refractory anemia; RARS, refractory anemia with ringed sideroblasts; RCMD, refractory cytopenias with multilineage dysplasia; 5q−, 5q minus syndrome; RAEB1, refractory anemia with excess blasts 1; RAEB2, refractory anemia with excess blasts 2; RAEB-t, refractory anemia with excess blasts in transformation; MDS-U, myelodysplatsic syndrome unclassified; RARS-T, refractory anemia with ringed sideroblasts with thrombocytosis; RCMD-RS, refractory cytopenias with multilineage dysplasia and ringed sideroblasts; AML, acute myelogenous leukemia; sAML, secondary AML; tAML, therapy-related AML; tMDS, therapy-related MDS; CMML, chronic myelomonocytic leukemia; PMF, primary myelofibrosis; VAF, variant allele frequency; CR, complete response; NR, no response; PR, partial response; PFS, progression-free survival; OS, overall survival; EFS, event-free survival; LFS, leukemia free survival; OR, odds ratio; ORR, overall response rate; mut, mutation; and WT, wild-type.
The strongest evidence for correlation of mutation status and response to therapy was found in patients carrying TET2 mutations. TET2 is one of the most common somatic mutations found in MDS (15%- 27%).25 TET2 encodes for a dioxygenase that converts 5-methylcytosine to 5-hydroxymethylcytosine leading to DNA demethylation. The link between methylation regulation and mechanism of action of HMA, azacitidine (AZA), and decitabine (DAC), has led a number of investigators to hypothesize mutations in this gene may affect response to these therapies. In fact, 3 large retrospective studies support improved response to hypomethylating therapy in patients with TET2 mutations.26-28 The first study to report an association by Itzykson et al27 evaluated 86 patients with MDS and AML with 20%-30% blasts that had been treated with AZA. Thirteen (15%) were found to carry the TET2 mutations (8 with MDS and 5 with AML). Compared with wild-type, patients in the TET2–mutated group had a higher incidence of lower hemoglobin, better cytogenetics and longer duration of MDS. After a median number of 6 cycles, response to AZA was increased in the mutated group with 9/13 (69%) responders compared with 23/73 (31%) in the wild-type (WT; p = 0.01). The complete response (CR) rate was similar between groups [mutated: 5/13 (38%), and WT: 15/73 (21%), p = 0.17]. In multivariate analysis, overall response rate (ORR) was positively affected by TET2 mutation, and negatively affected by poor-risk cytogenetics. Response was independent of hemoglobin, duration of MDS, or good-risk cytogenetics. Response duration and survival was not different between groups.
Two more recent studies by Traina et al28 and Bejar et al26 confirmed these initial observations. Traina et al28 evaluated 230 patients with MDS, MDS/MPN, or secondary AML, 92 (53 MDS, 28 MDS/MPN, 11 sAML) were treated with AZA or DAC between 2002 and 2010. Clinical characteristics that predicted for response were treatment with HMA therapy and platelet count ≥100. Direct sequencing of TET2 exons 3-11 was performed along with sequencing of DNMT3a, IDH1, IDH2, ASXL1, CBL, NRAS, KRAS, SF3B1, and TP53 as mentioned below. Forty-eight of 92 patients (52%) were previously untreated with HMA therapy. Focusing on the results of TET2 from this study, mutant TET2 occurred in 17 (18%) subjects. The numbers of responders was 5 (29%) and 12 (71%) nonresponders. This cohort included patients treated with AZA plus lenalidomide, which resulted in the highest response rate of 59% (10/17). The authors excluded this group from analysis and reported the response rate remained improved for TET2–mutated. In multivariate analysis, TET2 mutation, DNMT3a mutation, platelets, and WBCs were independent predictors of response. However, there was no association with PFS and OS in the TET2–mutated group. Unlike the Itzykson et al study,27 which only included high-risk MDS and sAML, this study also included low-risk MDS but still suggested improved responses in TET2–mutated patients to HMA therapy.
The final, and largest study, to evaluate TET2 mutation status and response to HMA therapy was by Bejar et al,26 who showed that the variant allele frequency (VAF), or the size of the malignant clone harboring the mutation, matters. They sequenced 40 genes in 213 MDS patients prior to treatment with HMA therapy from 2003 to 2010. Initial analysis showed that TET2–mutated patients showed no statistically significant increase in response (55% Mut vs 44% WT; p = 0.14) to HMAs. Because modern methods of sequencing for these mutations improve the sensitivity to be able to detect clone sizes of <1% of the population and can sequence across the entire gene in an unbiased manner, detection of patients with the mutation improves compared with traditional sanger sequencing of hot spots in a gene as was previously reported by Itzykson et al27 and Traina et al.28 The authors hypothesized that the sensitivity of their test was detecting small clones that would normally not be detected with traditional Sanger sequencing resulting in those patients being called wild-type. After setting a VAF threshold of >10%, they were able to demonstrate improved responses to therapy (60% Mut vs 43% WT; p = 0.036). This response was enhanced if co-occurrence of TET2 with ASXL1 is excluded. Once again, there was no association with OS. The observation that the size of the malignant clone harboring the potentially sensitizing mutation can affect response is an important one. This suggests the results obtained from commercial labs using hot spot Sanger sequencing of genes may be detecting a dominate clone as well as other minor clones which could affect individual patients response to hypomethylating therapy may not be detected but could be through deeper sequencing efforts. It also suggests that unless a mutation is in a substantial portion of malignant cells, any therapy (HMA or targeted) is unlikely to be associated with a clinical benefit. Finally, the authors found that mutations previously identified as associated with adverse prognostic significance (TP53, RUNX1, ASXL1, EZH2, or ETV6)15 in a patient cohort yet to be treated clinically with HMAs in practice, were no longer identified as predictive of shorter overall survival. This suggests that standard of care therapy with an HMA may abrogate some of the adverse prognostic impact of these mutations.
Another critical regulator of methylation is DNA (cytosine-5)-methyltransferase 3 alpha (DNAMT3a). DNMT3A encodes a DNA methyltransferase that catalyzes the incorporation of methyl groups to the cytosine residue of CpG dinucleotides. DNMT inhibitors can affect expression levels of DNMT1 and DNMT3a. DNMT3A is found recurrently mutated in MDS in 3%-13% of cases. Two studies previously discussed above by Traina et al28 and Bejar et al26 addressed response to therapy with HMAs and DNMT3a mutations. Earlier studies by Walter et al18 and Thol et al49 reported worse overall survival and shortened time to leukemic transformation but did not examine treatment response or had limited numbers of patients who had been treated with HMAs. Traina et al28 found an association of DNMT3a mutation and response, OR 3.59 (1.14-11.36; p = 0.03), in a multivariate analysis. There was no association with progression-free survival. Bejar et al26 also examined their cohort but found no association with DNMT3a mutation status and response to HMAs or survival. Therefore, there is no compelling evidence currently that DNMT3a mutations predict for or against response to HMA therapy in MDS.
Mutations in the spliceosome machinery genes are the most frequently found somatic mutations in MDS. They tend to be mutually exclusive but can co-occur with other somatic mutations such as those mentioned above. These mutations are thought to be early events in the generation of MDS. There is no evidence that mutations in these genes affects response to therapy but they are classically associated with a better overall prognosis.19,26,28-33 There are mixed associations with survival and mutations in spliceosome mutations along with leukemic transformation. Only 2 studies reported data on response to HMAs, Bejar et al26 and Traina et al28 only looked at SF3B1 by Sanger sequencing and found no association with treatment response. Bejar et al26 performed deep sequencing of known spliceosome factors recurrently mutated in MDS and found no association of mutation in any factor and response to hypomethylating therapy.
IDH1 and IDH2 mutations are uncommon in MDS (5%-10%). Isocitrate dehydrogenases are essential enzymes in the Krebs cycle and found to be recurrently mutated in a number of malignancies, resulting in accumulation of a metabolic by-product called 2-hydroxyglutarate (2-HG). 2-HG is an onco-metabolite that competitively inhibits prolyl hydroxylases and chromatin modifying enzymes, such as histone demethylases and TET-5mc hydroxylases.25 There is mixed data suggesting a correlation with response to therapy with HMAs.26,28,34-37 As above, older studies are limited by no information on treatment and a patient population studied prior to the advent of HMAs. Traina et al28 reported IDH1 and IDH2 mutations resulted in higher response rates to HMAs, but this was not found by Bejar et al.26 Because these are rare mutations in MDS, larger cohorts are needed to determine the impact of these mutations on therapy response.
TP53 mutations are rare in de novo MDS (5%-10%) and associated with complex cytogenetics. TP53 is more commonly mutated in treatment-related MDS and isolated del5q MDS. Three studies report no difference in response to HMAs in TP53–mutated patients.26,38,39 On the other hand, overall survival and progression to leukemia were shortened in TP53–mutated patients in all cohorts examined.
Other transcription factors associated with poorer prognosis in MDS, RUNX1, and ETV6,15 have no effect on response to hypomethylating therapy. In addition, recent evidence in a cohort of patients treated with hypomethylating therapy suggests that their adverse prognostic significance may not hold in the modern error of treatment with HMAs and transplant.26
Chromatin and histone modification machinery has also been implicated in MDS pathogenesis. The most commonly mutated are ASXL1 (10%-21%) and EZH2 (6%-8%).25 Both have been associated with poor clinical outcomes. There is no evidence that mutations in these genes affect response to hypomethylating therapy.26,28,40 Furthermore, as discussed above, the previously noted adverse prognostic impact of these mutations may be altered in the setting of treatment with HMAs.26
Finally, signal transduction can be altered in MDS. Two commonly mutated genes include CBL (1%-3%) and N-RAS (6%-17%).25 Mutations in CBL have been associated with lower response rates to HMAs,26 whereas NRAS mutations are not associated with response to therapy.26,41 Again because these are independent rare events in MDS, larger cohorts of patients will need to be studied to confirm or refute these findings.
Summary
The optimal use and timing of therapies for patients with high-risk de novo MDS remains an area of ongoing investigation. Current clinical prognostic scoring systems do not incorporate molecular genetic abnormalities in genes of histone/chromatin modification and DNA methylation that may predict response to HMAs. The choice of initial therapy in this case was treatment with 10 day decitabine on a clinical study and the patient achieved a complete response to treatment and subsequently underwent allogeneic transplant. The interactions of clinical factors, cytogenetic and molecular mutations is unknown but in this representative case the poor prognosis of t(3;21)42 may have been overcome by knowledge of TET2 mutation status and therapy with a HMA. Our review found no molecular mutations are strong predictors for resistance to therapies but response may be greater in TET2–mutated patients with high-variant allele frequencies. Based on the available evidence, we conclude that the choice of therapy between induction chemotherapy and HMAs based on molecular testing for recurrent somatic mutations is insufficient to recommend one over the other at the present time.
Differences in response and survival have not been demonstrated in randomized controlled trials. The overall grade of recommendation for choosing hypomethylating therapy over induction chemotherapy in high-risk MDS RAEB-2 based on molecular genetic mutations is grade 2C, and based on less-associated toxicity and increased responses primarily in TET2–mutated disease. Further prospective studies are needed to evaluate the long-term effects of hypomethylating therapy particularly in TET2–mutated patients.
Correspondence
Mark A. Schroeder, Washington University in Saint Louis, Division of Oncology, Section of BMT and Leukemia, 660 S. Euclid Ave, Campus Box 8007, St Louis, MO 63110; Phone: 314-454-8323; Fax: 314-454-7551; e-mail: mschroed@dom.wustl.edu.
