
Abstract
Complement is increasingly being recognized as an important driver of human disease, including many hemolytic anemias. Paroxysmal nocturnal hemoglobinuria (PNH) cells are susceptible to hemolysis because of a loss of the complement regulatory proteins CD59 and CD55. Patients with atypical hemolytic uremic syndrome (aHUS) develop a thrombotic microangiopathy (TMA) that in most cases is attributable to mutations that lead to activation of the alternative pathway of complement. For optimal therapy, it is critical, but often difficult, to distinguish aHUS from other TMAs, such as thrombotic thrombocytopenic purpura; however, novel bioassays are being developed. In cold agglutinin disease (CAD), immunoglobulin M autoantibodies fix complement on the surface of red cells, resulting in extravascular hemolysis by the reticuloendothelial system. Drugs that inhibit complement activation are increasingly being used to treat these diseases. This article discusses the pathophysiology, diagnosis, and therapy for PNH, aHUS, and CAD.
Learning Objectives
To understand the evidence supporting complement as a driver of hemolytic anemia
To describe the pathophysiology, diagnosis and treatment of atypical hemolytic uremic syndrome
Complement for the clinician
Complement activation
Complement is composed of >30 soluble and membrane-bound proteins and provides an important line of defense against bacteria, fungi, and viruses.1 It also facilitates the efficient removal of damaged cells and immune complexes. Activation of the complement cascade may be initiated by the following: (1) the lectin pathway triggered by carbohydrates found on bacteria; (2) the classical pathway triggered by immunoglobulin G (IgG) and IgM antibodies; (3) the alternative pathway that is constitutively activated through slow spontaneous hydrolysis of C3 (also referred to as tick-over); (4) thrombin that bypasses early pathways of complement activation by directly cleaving C3 and acting as a C5 convertase; and (5) plasmin and kallikrein that directly cleave C3 and its activation fragments (Figure 1). All of the activating pathways converge to form C3 convertases. Cleavage of C3 yields C3a and C3b, the latter of which triggers the formation of the C5 convertase. The C5 convertase cleaves C5 into C5a and C5b, the latter of which oligomerizes with C6, C7, C8, and multiple C9 molecules to form the membrane attack complex. Regardless of how the complement cascade is initiated, it has been estimated that the alternative pathway accounts for >75% of complement activation products.2 This is because the alternative pathway C3 convertase (C3bBb) not only triggers the C5 convertase; it also cleaves more C3 molecules leading to the rapid generation of C3b, resulting in an amplification loop.

Model of complement dysregulation in aHUS (A) and PNH (B). Initiators of the complement cascade (classical, alternative, and lectin pathway, as well as thrombin and plasmin/kallikrein) lead to C3 activation and C3 convertase formation, which is augmented the amplification loop (proximal complement). Consequently, membrane attack complexes (MAC) are formed on the membrane of target cells (terminal complement), leading to complement-mediated death. Production of C5a results in tissue factor and thrombin activation in a positive feedback loop. Complement dysregulation results from loss-of-function mutations in regulatory factors (Factor H, I, and THBD/thrombomodulin in aHUS and CD55, CD59 in PNH) shown in red, gain-of-function mutations (C3 and Factor B in aHUS) shown in green, and DGKE mutations in aHUS shown in gray, indicating the unknown effect on complement cascade. Eculizumab effectively inhibits terminal complement activation in both disorders by blocking the cleavage of C5 to C5a and C5b.
Model of complement dysregulation in aHUS (A) and PNH (B). Initiators of the complement cascade (classical, alternative, and lectin pathway, as well as thrombin and plasmin/kallikrein) lead to C3 activation and C3 convertase formation, which is augmented the amplification loop (proximal complement). Consequently, membrane attack complexes (MAC) are formed on the membrane of target cells (terminal complement), leading to complement-mediated death. Production of C5a results in tissue factor and thrombin activation in a positive feedback loop. Complement dysregulation results from loss-of-function mutations in regulatory factors (Factor H, I, and THBD/thrombomodulin in aHUS and CD55, CD59 in PNH) shown in red, gain-of-function mutations (C3 and Factor B in aHUS) shown in green, and DGKE mutations in aHUS shown in gray, indicating the unknown effect on complement cascade. Eculizumab effectively inhibits terminal complement activation in both disorders by blocking the cleavage of C5 to C5a and C5b.
The term complement infers that the complement cascade is an accessory of the immune system. Indeed, when Paul Ehrlich coined the term in 1899, its main function was thought to “complement” the effect of antibodies. It is now clear that the inability to regulate complement is an important driver of human disease and that preventing complement activation through highly targeted therapy can lead to dramatic clinical responses.
Complement regulation
Complement activation is tightly regulated by several membrane-bound and soluble complement regulatory proteins to protect bystander damage to host cells. A number of hemolytic disorders (Table 1) are caused by mutations and/or autoantibodies that inactivate complement regulatory proteins and mutations that directly activate the complement cascade. Most of these mutations and antibodies target the alternative pathway of complement (APC). Before discussing the individual complement-mediated hemolytic diseases, I will briefly discuss the complement regulatory proteins that are most relevant to human hemolytic diseases (Figure 1).
Pathogenesis of complement-mediated diseases
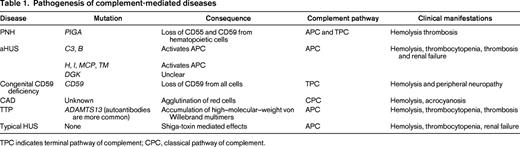
TPC indicates terminal pathway of complement; CPC, classical pathway of complement.
CD55 (decay accelerating factor) is a widely expressed glycosylphosphatidylinositol (GPI) anchor membrane protein that accelerates the decay of cell-surface–bound C3 convertases, thus limiting the formation of the C5 convertase and ultimately formation of the membrane attack complex. Because CD55 is a membrane protein, it only inhibits complement activation on cells that express it.
CD46 [membrane cofactor protein (MCP)] is a transmembrane cell-surface protein that also accelerates decay of C3 convertases. CD46 is expressed on most cell types but not erythrocytes. In conjunction with soluble factor I, CD46 also inactivates C3b to iC3b, thereby preventing reformation of the C3 convertase.
CD59 is another widely expressed GPI anchored membrane protein and is the major inhibitor of the terminal complement pathway. It functions by binding to C8 and C9 in the assembling membrane attack complex, thereby inhibiting pore formation of the membrane attack complex.
Factor I is synthesized in the liver and regulates the classical, alternative, and lectin pathways. In the alternative pathway, it cleaves and inactivates C3b.
Thrombomodulin is an endothelial cell receptor that modulates the generation of thrombin via its cofactor role in the activation of protein C; however, it also regulates factor I-mediated C3b inactivation.
Factor H regulates the alternative pathway in the fluid phase and on cell surfaces. It can bind directly to C3b and disrupt the C3 convertase of the alternative pathway. It also serves as an important cofactor for factor I to cleave and inactivate C3b.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired hemolytic anemia that results from the expansion of hematopoietic stem cells with a severe deficiency or absence of GPI, a glycolipid moiety that anchors more than a dozen different proteins to the cell surface of blood cells.3 In virtually all cases, GPI anchor deficiency in PNH results from a somatic mutation in PIGA, an X-linked gene whose product is required for the first step in GPI anchor biosynthesis. This results in the absence of complement regulatory proteins CD55 and CD59, leading to chronic complement-mediated hemolysis of the PNH erythrocytes. Hemolysis in PNH is chronic because of a continuous state of complement activation through tick-over, but paroxysms resulting in brisk hemolysis coincide with increases in complement activation that may occur with inflammatory states or surgery. The most common clinical manifestations include fatigue, dyspnea, hemoglobinuria, and abdominal pain,4 but thrombosis is the leading cause of death in PNH.5
GPI biosynthesis is a posttranslational event that occurs in the endoplasmic reticulum.6 There are >10 steps and >30 gene products required. The PIGA gene product is 1 of 7 proteins involved in the first step of GPI anchor biosynthesis. Theoretically, a mutation of any gene in the pathway could lead to PNH; however, most cases are caused by PIGA mutations. This is because PIGA is on the X chromosome; therefore, a single mutation in a hematopoietic stem cell will result in GPI anchor protein deficiency (males have a single X chromosome and females have only one active X chromosome because of lyonization). The remaining known genes in the GPI anchor biosynthethic pathway are on autosomes; thus, 2 “hits” disrupting function on both alleles would be necessary to interrupt GPI anchor synthesis. Recently, a PNH patient was demonstrated to have a compound heterozygous mutation in PIGT.7 He was born with a heterozygous germline splice site mutation on one allele and acquired an 8-Mb deletion involving the other PIGT allele, which abolished the expression of GPI anchored proteins, resulting in intravascular hemolysis and smooth muscle dystonias.
The absence of CD59 is most responsible for the clinical manifestations in PNH. Accordingly, rare cases of inherited mutations in CD59 leading to loss of CD59 on the cell surface have been well documented.8,9 The phenotype of these patients mimics PNH in that they manifest chronic intravascular hemolysis with paroxysmal flares of hemolysis and a propensity for thrombosis. Unlike PNH, pedigrees with inherited CD59 deficiency also present with relapsing immune-mediated peripheral neuropathy. In classical PNH, the CD59 deficiency is only found on the blood cells; in patients with germline CD59 mutations, CD59 is deficient in all cells in the body. Thus, germline CD59 deficiency may be associated with demyelination via activation of terminal complement.
Eculizumab is a humanized monoclonal antibody that blocks terminal complement by binding to C5 and is the only Food and Drug Administration (FDA)-approved therapy for PNH.10-13 It is also effective in patients with PNH resulting from germline CD59 deficiency and compound heterozygous mutations in PIGT.7,14 The drug is administered intravenously every 7 days for the first 5 weeks and then biweekly thereafter. Eculizumab inhibits the formation of the membrane attack complex and in doing so compensates for the CD59 deficiency of PNH patients. It does not compensate for the CD55 deficiency; thus, eculizumab is highly effective in abrogating the intravascular hemolysis in PNH, but most PNH patients on eculizumab will continue to experience mild-to-moderate extravascular hemolysis attributable to C3d deposition on the PNH red cells.15 This explains why >50% of PNH patients treated with eculizumab develop a positive direct antiglobulin test (C3 positive but IgG negative) in conjunction with a mild-to-moderate anemia and elevated reticulocyte count.16 The most serious risk of terminal complement blockade is life-threatening Neisserial infections (∼0.5%/year). Thus, all patients treated with eculizumab should be vaccinated against Neisseria.
The majority of classical PNH patients will respond to eculizumab; however, the hemoglobin response is highly variable and may depend on underlying bone marrow failure, concurrent inflammatory conditions, genetic factors, and the size of the PNH red cell clone after therapy.17 In fact, 25% to 35% of patients continue to require red cell transfusions despite treatment with eculizumab. The most common reason for continued transfusions is extravascular hemolysis. An increase in the percentage of PNH red cells after eculizumab therapy correlates with response but also with extravascular hemolysis. The PNH red cells that are protected by eculizumab are CD55 deficient and thus are susceptible to opsonization by C3 fragments and premature removal in the spleen.15 Interestingly, up to 10% of PNH patients treated with eculizumab have a decrease in the percentage of PNH erythrocytes despite a large percentage of PNH granulocytes.17 These patients achieve normal hemoglobin levels without the aid of transfusions. Pharmacogenetics has also been shown to influence response to therapy. Polymorphisms in the complement receptor 1 (CR1) gene are associated with response to eculizumab. CR1, through binding C3b and C4b, enhances the decay of the C3 and C5 convertases.18 The density of CR1 on the surface of red cells modulates binding of C3 fragments to the GPI-negative red cells when C5 is inhibited. PNH patients with polymorphisms in CR1 that lead to low CR1 levels (L/L genotype) are more likely to be suboptimal responders to eculizumab than patients with intermediate (H/L genotype) or high (H/H genotype) levels of CR1. More recently, it has been discovered that a single missense C5 heterozygous mutation, c.2654G → A, prevents binding and blockade by eculizumab while retaining the functional capacity to cause hemolysis.19 The c.2654G → A polymorphism is present in 3.5% of the Japanese population and accounts for the poor response to eculizumab in patients who carry the mutation.
Indications for terminal complement inhibition
Eculizumab is expensive and must be administered indefinitely for a sustained response; thus, in patients with mild or no symptoms, I simply follow them with a watchful waiting approach regardless of the size of the PNH clone. Severe anemia, thrombosis, frequent pain paroxysms, debilitating fatigue, worsening renal insufficiency, or dyspnea are good indications to initiate therapy.20 Therapeutic decisions should not be based solely on the PNH clone size; however, patients with a large clone (>50% PNH granulocytes and >10% PNH red cells) coupled with a markedly elevated lactate dehydrogenase (LDH; indicator of intravascular hemolysis) and a robust reticulocyte count (indicator of adequate bone marrow reserve) are most likely to benefit. Eculizumab does not improve bone marrow failure. Patients with ongoing bone marrow failure from aplastic anemia (hypocellular bone marrow, severe thrombocytopenia, and a relatively low reticulocyte count) are less likely to derive benefit from eculizumab. In these patients (aplastic anemia with a small PNH clone), it is best to direct initial therapy toward the bone marrow failure.
Monitoring the PNH patient on eculizumab
I monitor patients on eculizumab with a complete blood count, reticulocyte count, LDH, and biochemical profile weekly for the first 4 weeks and then monthly thereafter (Table 1). A direct antiglobulin test should be obtained in patients with evidence of persistent hemolysis, and PNH flow cytometry should be obtained yearly because the PNH clone size may fluctuate over time. The LDH usually returns to normal or near normal within days to weeks after starting eculizumab; however, the reticulocyte count usually remains elevated and the hemoglobin response is highly variable. The reticulocyte count often remains elevated because most PNH patients on eculizumab continue to have extravascular hemolysis because of deposition of C3 fragments on PNH red cells. The hemoglobin response is primarily dependent on the degree of extravascular hemolysis and the amount of underlying bone marrow failure.
In classical PNH patients who are transfusion dependent, a marked decrease in red cell transfusions is usually observed, with >70% achieving transfusion independence.21 Breakthrough intravascular hemolysis and a return of PNH symptoms occurs in <5% of PNH patients treated with eculizumab. This typically occurs 1 or 2 days before the next scheduled dose and is accompanied by a spike in LDH levels. If this occurs regularly, the interval between dosing can be shortened to 12 days, or the dose of eculizumab can be increased to 1200 mg, every 14 ± 2 days. Increased complement activation may accompany infections (upper respiratory viruses, influenza, viral gastroenteritis, etc.) or trauma and may result in transient breakthrough hemolysis. These single episodes of breakthrough hemolysis do not require a change in dosing because patients usually return to their baseline hemoglobin once the inflammation has resolved.
Atypical hemolytic uremic syndrome
Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy (TMA) that presents with intravascular hemolysis, thrombocytopenia, and acute renal failure.22 Most cases of aHUS are caused by mutations in genes encoding proteins involved in the APC or by autoantibodies directed against APC regulatory proteins.23 These mutations can effect proteins that help degrade cell-surface C3b (APC inhibitors), such as factor H (most common), factor I, MCP, and thrombomodulin or proteins that drive the APC (activating mutations) in C3 or factor B. Rare cases of early onset (<12 months of age) aHUS harbor mutations in diacylglycerol kinase-ε (DGKE); however, the mechanism of how DGKE mutations cause aHUS is unclear.24
aHUS is a two-hit disease
In most instances, aHUS appears to be a “two-hit” disease. This is highlighted by the familial form of the disease in which the penetrance is ∼50%.25 In most patients, there is a putative “trigger” (pregnancy, infection, surgery, etc.) that leads to inflammation and presumably increases complement activation. The second hit may also be in the form of an autoantibody or a genetic modifier; ∼20% of patients have mutations in more than one gene, and some patients with factor H mutations also have autoantibodies to factor H. Most patients with autoantibodies against factor H have a mutation that abolishes expression of complement factor H-related protein-1 (CFHR1).26 Recent structural data suggest that the CFHR1 mutation may predispose to autoantibody formation by inducing a neoepitope.
Differentiating aHUS from other thrombotic microangiopathies
aHUS presents with TMA and widespread end-organ damage that classically involves the kidneys; however, aHUS often presents with end-organ damage that can involve the brain, gastrointestinal tract, liver, and heart. Consequently, clinical manifestations alone are not reliable in distinguishing aHUS from typical HUS or thrombotic thrombocytopenic purpura (TTP). In patients presenting with TMAs, it is important to obtain ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motifs, member 13) levels and screen for Shiga toxin before instituting definitive therapy. Plasma exchange is often initiated before the results of these assays return because of the aggressiveness of TMAs. If ADAMTS13 activity is <10%, a diagnosis of TTP is established; the presence of an inhibitor to ADAMTS13 establishes that the disorder is acquired and not hereditary. If ADAMTS13 activity is >10% and Shiga toxin assay is negative, a diagnosis of aHUS must be considered.22 Unfortunately, there is no definitive test to make a diagnosis of aHUS, and therapy is often delayed or not administered given the high cost of eculizumab. Genetic testing for mutations that lead to increased activation of APC is expensive, takes several weeks to return, and is only informative in ∼50% to 60% of cases (Figure 2).23,27 Frequently, genetic variants of unknown significance are identified, providing clinicians with data of uncertain utility.28,29 Biomarkers of APC such as C5a or soluble C5b-9 have been compared in aHUS and TTP, but these are not always reliable in distinguishing the two diseases.30 Our laboratory has recently described a modified (indirect) Ham test to differentiate aHUS and TTP.31 The principle of the “indirect Ham test” is similar to the acidified serum (Ham) test that was used to diagnose PNH before the routine use of flow cytometry (Figure 3). In PNH, there is a genetic mutation, PIGA, that causes a loss of complement regulatory proteins (CD55 and CD59) on red cells. Acidifying normal human serum activates APC and leads to specific lysis of PNH erythrocytes because they are unable to protect themselves from the activated complement in acidified serum. In most aHUS cases, there are genetic mutations or antibodies that lead to activation APC in the patient's serum. Thus, when PNH-like reagent cells (PIGA null TF-1 cells) are incubated with aHUS serum, they rapidly accumulate C5b-9 and undergo cell death within hours. Complement-induced death prevents conversion of WST-1 into formazan, a red dye (Figure 3B). Although high–molecular–weight multimers of von Willebrand factor can also activate the APC,32 the degree of APC activation in aHUS is much higher than that of TTP, allowing distinction between the two diseases in this new assay. Our laboratory was able to demonstrate APC activation in aHUS serum regardless of whether the patient's serum was studied in the acute phase or in remission, highlighting the requirements for two hits to cause the disease (Figure 3B). This assay will need to be validated in a larger patient cohort before routine use is recommended. Furthermore, because it is a serum-based assay, it may not detect the 10% to 15% of aHUS patients with membrane mutations such as MCP, thrombomodulin, or DGKE.

Frequency of genetic lesions in aHUS. CFH indicates complement factor H; CFI, complement factor I; THBD, thrombomodulin.
Frequency of genetic lesions in aHUS. CFH indicates complement factor H; CFI, complement factor I; THBD, thrombomodulin.

A) Model of Ham test for PNH (top) and indirect Ham test for aHUS diagnosis (bottom). The Ham test evaluates the effect of acidified serum on patient's cells, whereas the modified test evaluates the effect of patient's serum on GPI-anchored protein–deficient reagent cells. Both tests use absorbance changes as readouts of cell viability. GPI-AP indicates glycosylphosphatidylinositol-anchored proteins. B) WST-1 cell viability assay in patients with aHUS. Percentage of nonviable reagent cells in aHUS after exposure to acute-phase serum, remission serum on eculizumab (Ecu), and remission serum off eculizumab compared with acute or remission serum from TTP patients.
A) Model of Ham test for PNH (top) and indirect Ham test for aHUS diagnosis (bottom). The Ham test evaluates the effect of acidified serum on patient's cells, whereas the modified test evaluates the effect of patient's serum on GPI-anchored protein–deficient reagent cells. Both tests use absorbance changes as readouts of cell viability. GPI-AP indicates glycosylphosphatidylinositol-anchored proteins. B) WST-1 cell viability assay in patients with aHUS. Percentage of nonviable reagent cells in aHUS after exposure to acute-phase serum, remission serum on eculizumab (Ecu), and remission serum off eculizumab compared with acute or remission serum from TTP patients.
Treatment of aHUS
Plasma exchange is usually the initial therapy for aHUS because of the difficulty in differentiating aHUS and TTP, the time it takes to get the results of ADAMTS13 activity, the expense of eculizumab, and the fact that aHUS is often a diagnosis of exclusion. Once plasma exchange is initiated, it is often hard to discontinue because it is common to see early improvements in the LDH levels and platelet counts; however, end-organ complement-mediated damage often persists. After 3 years of follow-up, end-stage renal disease or death occurs in >50% of aHUS with or without plasma-based therapy.23 Eculizumab is the only FDA-approved drug for the treatment of aHUS based on two open-label, prospective trials in a total of 37 patients.33 During treatment, 31 patients achieved a TMA event-free status. More than half of the patients achieved improved renal function and some were able to discontinue dialysis. Thirty-three patients enrolled in a 2-year extension trial, and, after completion, all were alive and TMA free. Platelet and LDH response tend to occur within a few days to 1 week; renal, central nervous system, and other end-organ damage can take longer. It is unclear whether or not responding patients need to remain on eculizumab. More recent studies suggest that at least some patients can discontinue the drug after they achieve complete remission.31 If a diagnosis of aHUS is made early in the disease course, eculizumab may be cost-effective in reducing the duration of hospitalization, multiple weeks of plasma exchange, and the morbidity/mortality of the disease.
Cold agglutinin disease
Cold agglutinin disease (CAD) is classified as either primary or secondary. Primary CAD is a clonal B cell lymphoproliferative disorder that is called “primary cold agglutinin-associated lymphoproliferative disease”; it is distinct from lymphoplasmacytic lymphoma (MYD88 L265P negative), marginal zone lymphoma, and other low-grade lymphoproliferative diseases.34 Secondary CAD is a syndrome associated with a variety of infectious and neoplastic disorders (aggressive lymphomas, Hodgkin's lymphoma, carcinomas, etc.). Cold agglutinins are autoantibodies (typically IgM) that agglutinate red blood cells at 4°C but may also act at warmer temperatures. In the workup of a cold agglutinin, it is important to ascertain the thermal amplitude and the titer. Most pathogenic cold agglutinins are of the IgM class, have a thermal amplitude that exceeds 28 to 30°C, and have a titer of 1:256 or greater. In contrast, nonpathogenic cold agglutinins are low titer and have low thermal amplitude.
Mechanism of hemolysis in CAD
Cold agglutinins with relatively high thermal amplitude bind to red cells in acral parts of the circulation (e.g., fingers, toes, ears, etc.) and are commonly directed against the I antigen. Binding of IgM-CA activates the classical complement pathway. C1 esterase activates C4 and C2, ultimately generating the C3 convertase, which cleaves C3 to C3a and C3b. After return to central portions of the circulation (∼37°C), the IgM-CA dissociates from the cell surface but C3b remains bound to the cell. The C3b-coated red cells are then sequestered by macrophages of the reticuloendothelial system, predominantly in the liver (extravascular hemolysis). C3b of the surviving red cells is eventually cleaved, leaving a high number of circulating red cells with C3d on the surface. This explains why patients with CAD have a direct antiglobulin test that is strongly positive for C3d but negative for IgG and IgM. In most cases of CAD, the classical pathway activity does not proceed to activate the terminal pathway of complement attributable to the expression of CD55 and CD59 on the red cell membrane; nevertheless, in ∼10% of patients, usually in association with a severe exacerbation (infection, surgery, etc.), intravascular hemolysis resulting from terminal activation may occur.
Treatment of CAD
Patients with primary CAD should be instructed to avoid cold temperatures if possible and especially to keep their hands, feet, and ears protected in cold environments; however, symptomatic patients will often need pharmacologic therapy. Rituximab is the best first-line therapy.35 It leads to remission (median duration of 1 year) in ∼50% of patients. Relapsed patients may respond to subsequent rituximab therapy. Fludarabine/rituximab combination therapy has a 75% response rate (median duration over 5 years) but significantly more toxicity.36 Rituximab and bendamustine combination therapy and bortezomib-based therapy also has activity in primary CAD. There are case reports of eculizumab therapy in the occasional CAD patient with intravascular hemolysis, and there is in vitro evidence that a monoclonal antibody that targets C1s can help to control extravascular hemolysis.37 However, these drugs have not been approved. In patients with secondary CAD, treatment should target the underlying cause.
Summary
Complement has been known to play a pathophysiologic role in human disease since the middle of 20th century; however, in the past decade, it is being recognized as a major driver of numerous blood diseases and as an important drug target. Research into developing novel complement inhibitors is underway, and, in the next decade, the role for complement inhibition is likely to expand.38-40
Correspondence
Dr Robert A. Brodsky, Division of Hematology, Department of Medicine, Johns Hopkins University School of Medicine, 720 Rutland Ave, Ross Research Bldg, Rm 1025, Baltimore, MD 21205-2196; Phone: 410-502-2546; Fax: 410-955-0185; e-mail: rbrodsky@jhmi.edu.
