
Abstract
Peripheral T-cell lymphomas (PTCL), with the exception of anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma (ALCL), have a very poor prognosis. Although current first line chemotherapy continues to be a CHOP-like (cyclophosphamide, doxorubicin, vincristine, prednisone) regimen there is now data suggesting that the addition of etoposide in younger patients improves outcomes. Even for those patients who do have a response to therapy, the risk of relapse remains quite high. Although autologous transplant in first remission is often used, its role as consolidation therapy in first remission remains unclear and may preferentially benefit low-risk patients. In the relapsed setting, major advances have occurred with Food and Drug Administration (FDA) approval of 4 new agents (pralatrexate, romidepsin, belinostat, brentuximab vedotin) for relapsed/refractory PTCL since 2009. These 4 drugs represent the first agents ever approved specifically for this indication. Unfortunately, with the exception of ALCL for which brentuximab vedotin will likely substantially change our approach to treatment, there are still many patients for whom available drugs will not be effective, and it is for these patients that further advances are urgently needed.
Learning Objectives
To appreciate the poor prognosis of peripheral T-cell lymphoma, not otherwise specified, (PTCL, NOS), and superior outcomes for anaplastic lymphoma kinase + (ALK+) anaplastic large cell lymphoma (ALCL)
To understand the exciting new drugs that will further improve outcomes in ALCL
To learn about ongoing questions concerning the best therapeutic approach in PTCL
The 22 distinct subtypes of peripheral T-cell lymphoma (PTCL) represent a heterogeneous and complex group of lymphomas that constitute 7% of non-Hodgkin's lymphoma and span the spectrum of outcome from the most lethal lymphomas to 5 year survival rates in the 70% range. In North America, PTCL-NOS is the most common form of peripheral T-cell lymphoma comprising 34% overall of T-cell lymphomas with ALCL-ALK+ at 16% and ALCL-ALK− at 7.8%. On the Asian continent, PTCL-NOS comprises 22% of T-cell lymphomas with ALCL-ALK+ at 3.2% and ALCL-ALK− at 2.6%.3
The PTCL, NOS, is a heterogeneous group of T-cell lymphomas that do not fit into any of the specific subtypes defined by the World Health Association classification system.4 The malignant cells are CD4+/CD8− with frequent loss of CD5 and CD7 and are usually medium to large in size with high proliferation rates. T-cell receptor beta chains are typically clonally rearranged. The karyotype is typically complex with a wide variety of chromosomal abnormalities. Median age at presentation is 57 years and most patients have advanced stage disease at diagnosis (69%) with frequent extranodal involvement of the skin (16%), liver (12%), and bone marrow (21%).2
ALCL is a CD30+ T cell neoplasm. Morphologically cells can be large to small but the classic “hallmark cell” is always present and has an eosinophilic region near an eccentric, kidney or horseshoe shaped nuclei. ALCL is divided into ALK+ and ALK−, which have drastically different prognosis with 5 year overall survival of 70% versus 49%, respectively.2 With rare exceptions, ALK is absent in normal human tissue, but immunohistochemistry shows positive ALK staining in the cytoplasm and nucleus of most ALK+ ALCL. The increased ALK expression is due to a translocation, usually t(2;5)(p23;q35), which fuses the ALK gene on chromosome 2 with the nucleophosin gene on chromosome 5 resulting in constitutive activation of ALK, a tyrosine kinase receptor that is part of the insulin superfamily. ALK+ and ALK− ALCL are heterogeneous diseases and the vastly different prognoses may be at least partially due to their different clinical presentations. ALK+ ALCL more commonly occurs in children and young adults with a median age of 34 years versus 58 years in ALK- ALCL. Similar to PTCL, NOS, ALCL ALK− patients frequently have advanced stage disease with the involvement of extranodal sites and the presence of B symptoms at the time of diagnosis.
Treatment options for PTCL span the spectrum from single-agent immunosuppression in AITL to allogeneic stem cell transplant. In the largest reported series of PTCL-NOS, 5 year overall survival is 32% and 5 year failure-free survival is 20%.3 Despite these discouraging outcomes, 10 year overall survival may be as high as 20% suggesting a small group of patients may be cured. The optimal treatment regimen at diagnosis and relapse is unclear since the data comes largely from small studies, which include a heterogeneous group of lymphoma subtypes and a variety of different chemotherapy regimens which may or may not include stem cell transplant. Analyses of these data indicate that anthracycline-based regimens, particularly CHOP, are most commonly used and that the addition of etoposide in special subgroups of younger individuals may improve outcome.
Historically treatment of PTCL and ALCL has been difficult and outcomes have been poor. Now, a better understanding of subtypes, development of new therapies, and using drugs with new mechanisms of action are changing the approach to these diseases.
Outcomes and prognosis
The International Peripheral T-Cell Lymphoma Project is a collaborative effort of 22 institutions in North America, Europe, and Asia. This series of newly diagnosed patients with PTCL, who had pathology that underwent central expert review, provides a wealth of data on PTCL. Of the 1314 patients, 331 had PTCL-NOS and 159 patients had systemic ALCL (ALK+, 87; ALK−, 72).2 The data listed in Table 1, document the better overall survival for ALK+ ALCL at 70% compared with ALK− ALCL at 49% and PTCL-NOS at 32%.
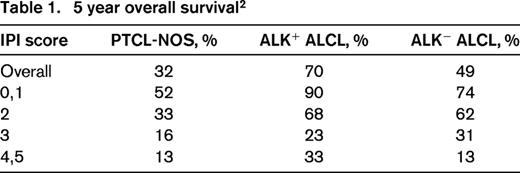
Although we are used to thinking about PTCL as a very poor survival group of diseases, the data in Table 1 and the survival curves in Figure 3 reaffirm that the International Prognostic Index (IPI) score differentiates between higher-risk and lower-risk patients with PTCL.2 One-half of patients with PTCL, NOS, who have a low risk IPI of 0/1 will be alive in 5 years, and conversely, even ALK+ ALCL patients don't do well if they have a high-risk IPI of 4/5 with 5 year overall survival (OS) of only 33%.
The Intergruppo Italiano Linfomi retrospectively analyzed 385 patients with PTCL, NOS in order to develop a Prognostic Index for T-cell Lymphoma (PIT).5 Four factors were found to be independent in multivariate analysis: age >60; performance status ≥ 2; lactate dehydrogenase (LDH) > upper limits normal; and bone marrow positive. Using these factors they were able to separate patients into 4 risk groups with 5 and 10 year OS of 62.3% and 54.9% for group I (0 factors), 52.9% and 38.8% for group II (1 factor), 32.9% and 18% for group III (2 factors), and 18.3% and 12.6% in group IV (3-4 factors). The major difference between the IPI and the PIT is in the PIT's ability to separate the outcomes for the intermediate risk groups, although the curves for the low-intermediate and the high-intermediate risk groups on the IPI survival curve are almost superimposable. An examination of the survival curves in Figure 1 leads to 3 conclusions concerning PTCL-NOS.1 First, failure-free survival (FFS) is poor with a median FFS <1 year. Second, there is a slow steady rate of relapse after year 5 resulting in a ∼10% rate of relapse in this 10 year period. Third, 20% of the patients are alive at year 15. Ttogether, these observations support 3 types of progression: a very rapid early progression of 50% per year; a middle phase with 20% recurrence in years 1-6; and a late phase with 10% recurrence over years 7-14. What is also of interest is that even when the overall survival is divided by IPI, these same 3 phases apply. Understanding the biology of these three phases may well lead to treatments directed not by prognostic factors but by the biology of these diseases.

The failure free survival curves in Figure 2 show that these same 3 concepts can be applied to ALK− ALCL.2 First, the 5 year FFS is better but still not good at 36%. Second, after 4 years there are no relapses. Third, 36% (estimated from Figure 2B) of the patients are alive at year 15.


(A) Overall survival by IPI for ALK+ ALCL. (B) Overall survival by IPI for ALK− ALCL.2
Again applying these same concepts to ALK+ ALCL, first, FFS at 5 years is initially better at 70%.2 Second, the rate of relapse decreases but continues indefinitely. Third, ∼60% (estimated from Figure 2B) of the patients are alive approaching year 15.
When looked at from the perspective of these long-term survival curves, PTCL-NOS and ALK+ ALCL and ALK− ALCL are very different diseases. PTCL-NOS has a very high early mortality but there are patients who then progressively relapse over a decade and some who relapse very late. ALK+ ALCL has excellent early survival but a continuing recurrence rate indefinitely. ALK− ALCL has worse early survival but virtually no relapses after year 4.
Current standard of care for initial therapy for PTCL
Induction chemotherapy
PTCL that is primarily refractory to chemotherapy occurs in up to 30% of patients.3 Therefore, achieving remission is one of the first challenges to curing PTCL, NOS, and ALCL. An analysis of studies completed over the last 20 years demonstrates how little progress we have made in the past 20 years. The first study performed specifically in PTCL was of VACPE, a CHOP like regimen combined with etoposide which was published in 1996.6 A complete response (CR) rate of 75% was achieved in patients with PTCL resulting in a 5 year overall survival (OS) of 48% and disease-free survival (DFS) of 62%. Unfortunately, to date, not much progress has been made and the current standard of care in many patients continues to be a CHOP-like regimen plus etoposide.
Current recommendations are based on an analysis by the German High Grade Non-Hodgkin Lymphoma Study Group (DSHNHL) of the outcomes of 289 patients with PTCL who were enrolled in clinical trials comparing CHOP± etoposide in patients with aggressive B- and T-cell lymphomas.7 Their data showed that patients who were ≤60 years old and had a normal LDH at diagnosis had significantly improved 3 year event-free survival (EFS) with the addition of etoposide to CHOP (51.0% vs 75.4%; p = 0.03) although there was no difference in OS (p = 0.176). The significant benefit from etoposide was predominantly seen in the good prognosis ALK+ ALCL lymphoma group with a 3 year EFS of 57.1% without versus 91.2% with etoposide (p = 0.012); there was, however, a trend toward improved 3 year EFS in the other subtypes, 60.7% versus 48.3% (p = 0.057). It is one of the only studies that has shown any improvement over CHOP and provides the basis for incorporating etoposide into chemotherapy regimens for younger patients.
Relapses are frequent resulting in a 3 year progression-free survival (PFS) in some reports as low as 32%.8 Therefore, the second major challenge to curing PTCL is maintaining the remission. First line consolidation with stem cell transplant (SCT) has been utilized by many centers in an attempt to prolong remissions and improve survival. Unfortunately, no large randomized trials have been performed to assess the benefit of SCT versus standard chemotherapy in the upfront setting. Furthermore, the data available from retrospective case series or small non-randomized clinical studies is difficult to interpret because of the heterogeneity of patients, pathology, and treatment regimens (Table 2). These studies are typically skewed in favor of SCT by inclusion of the good risk ALK+ ALCL and by exclusion of the poor risk chemo-refractory patients who could not proceed to transplant. So, although the data consistently show that patients who get to transplant survive longer than those who do not (3 year OS of 71% vs 11%),9 this may be a measure of underlying biology rather than because of a specific therapeutic intervention.
Prospective studies of ASCT in first remission
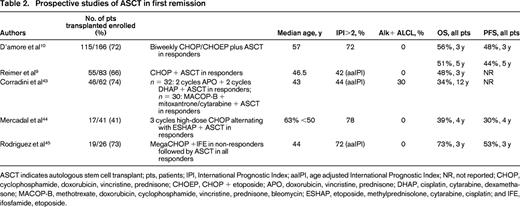
ASCT indicates autologous stem cell transplant; pts, patients; IPI, International Prognostic Index; aaIPI, age adjusted International Prognostic Index; NR, not reported; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; CHOEP, CHOP + etoposide; APO, doxorubicin, vincristine, prednisone; DHAP, cisplatin, cytarabine, dexamethasone; MACOP-B, methotrexate, doxorubicin, cyclophosphamide, vincristine, prednisone, bleomycin; ESHAP, etoposide, methylprednisolone, cytarabine, cisplatin; and IFE, ifosfamide, etoposide.
The largest prospective phase II trial of autologous SCT in first remission (NLG-T-01) included 166 patients with PTCL, excluding ALK+ ALCL, with the intent to transplant all patients who had ≥ partial response (PR) to chemotherapy.10 Initial therapy was biweekly CHOP in patients >60 and CHOEP in younger patients. The overall response rate (ORR) and complete response/complete response unconfirmed (CR/Cru) rates were 82% and 51%, respectively, so that 72% of enrolled patients were able to undergo transplant. The 5 year PFS and OS for all patients, transplanted or not, were 44% and 51%, respectively. For the ALK- ALCL patients the PFS and OS were 61% and 70%, whereas for the PTCL, NOS, patients it was 38% and 47%, demonstrating again that ALCL, even when Alk-negative, has a superior result compared to the other PTCLs.
The Nordic study did not provide us with definitive proof that patients do better with SCT than with conventional chemotherapy, although it did show that there are a group of patients who are long-term survivors with this approach. However, with the caveat that cross trial comparisons are fraught with bias, it is disheartening that the 5 year OS of 51% seen in the Nordic transplant trial in 201210 is so similar to the 48% reported in the VACPE trial in 1996.6
Because of the lack of solid clinical trial data and the understanding that some patients will be cured even without transplant, physicians are divided on whether or not to offer consolidative transplant to PTCL patients in first remission. The only area where there is general agreement is that patients with low risk alk+ ALCL should not be considered for first remission transplant because of their excellent prognosis with standard therapy. A large, well-designed randomized control trial is needed, but T-cell non-Hodgkin lymphoma (NHL) is so rare that we are unlikely to ever have a definitive answer.
Management of relapsed disease
Progress had been made in the management of relapsed PTCL with the approval of 4 new agents for this indication (Table 3).11-17 In addition to the drugs FDA approved for relapsed/refractory PTCL, there is one additional drug with phase II data and 6 recommended combinations, the details of which are reported in Table 3. The importance of these drugs lies in the fact that responses may be durable in a minority of patients who would then become eligible for stem cell transplant and potential long remissions.
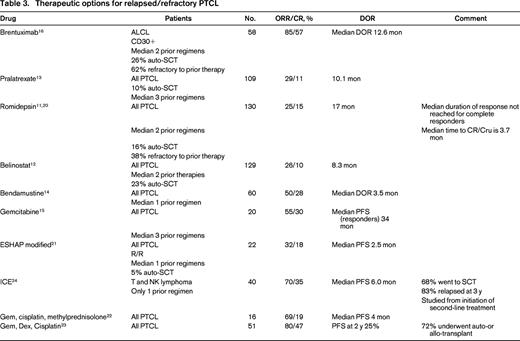
Brentuximab vedotin (BV) is an anti-CD30 monoclonal antibody fused to an anti-tubulin agent, monomethyl auristatin E (MMAE), by a protease cleavable linker. The anti-CD30 component allows the drug antibody to bind to the CD30 receptor and then be ingested into the cell where the anti-tubulin agent, MMAE, binds to tubulin and disrupts the tubulin structure, leading to cell cycle arrest and cell death. The ORR was 85% with a complete response rate of 57% and a duration of response (DOR) of 12.6 months in responders and 13.2 months in complete responders.16 Remarkably, over 50% of the patients had not progressed at 12 months. The grade 3-4 toxicities occurring in >10% of patients were neutropenia 21%, thrombocytopenia 14%, and peripheral sensory neuropathy 12%. These kinds of results support the bridge to transplant concept, which can lead to even more durable responses.
Brentuximab vedotin was initially studied and approved specifically for use in relapsed/refractory systemic ALCL, all of which are CD30-positive. However, research is ongoing into its role in other PTCLs, as CD30 protein expression has been demonstrated in 58% of PTCL, NOS, patient samples.18 Thus far, although responses in other T-cell lymphoma subtypes have not been as robust as those seen in ALCL, they are still encouraging with ORR 33% (CR 14%) in 21 patients with PTCL-NOS.19 Its role in the management of other CD30+ PTCL is a topic of continued investigation.
Pralatrexate was the first drug ever FDA approved for use in relapsed/refractory PTCL. It is an antifolate drug designed to have high affinity for the reduced folate carrier to increase internalization of drug; furthermore, because it is effectively polyglutamylated it is minimally extruded. The 115 patients enrolled in this phase II trial is small number for a trial that led to FDA approval of a new drug; however, in this rare disease, larger trials are generally not feasible and most of the trials that have led to drug approval for T-cell lymphomas have had a similar number of subjects. Patients in this study were heavily pretreated with a median of 3 prior treatments. One hundred eleven patients received study drug, but 2 were not evaluable for response because their PTCL diagnosis could not be confirmed. The median ORR for all patients was 29% with a CR/CRu rate of 11% and median DOR of 10.1 months.13 The ORR in PTCL-NOS was 32%. More than 25% of the patients remained progression free at 12 months. Although the median time to response was only 46 days, the median time to best response was much longer at 141 days, so patients who are tolerating drug should be continued on drug for several cycles before declaring lack of efficacy.
The grade 3-4 adverse events >10% were thrombocytopenia 32%, neutropenia 22%, and anemia 18%. Grade 3-4 mucositis occurred in 22% of patients despite folate and vitamin B12 supplementation. In hopes of making pralatrexate a more tolerable drug, there is an ongoing phase II trial examining the use of the antifolinic acid, leucovorin, in combination with pralatrexate (NCT02106650) to evaluate for frequency and severity of mucositis.
Romidepsin was approved by the FDA on November 5, 2009, for relapsed/refractory PTCL. This selective class I histone deacetylase inhibitor (HDAC-I) increases histone acetylation, which results in increased tumor suppressor activity, growth inhibition, and effects on the cell cycle leading to apoptotic death. The phase II, non-randomized trial, enrolled 130 evaluable patients with relapsed/refractory PTCL.11 Although the ORR of 25% and CR/CRu of 15% were low, longer follow-up has shown a median DOR of 28 months (range, <1-48+) for all responders and a median that has not been reached for those who achieved a CR/CRu.20 The grade 3-4 toxicities >10% were thrombocytopenia at 23% and neutropenia at 18%. The data for the 69 patients with PTCL-NOS were ORR of 25% and CR/Cru of 14%.
Belinostat is a pan-histone deacetylase inhibitor inhibiting Classes I, II, and IV HDAC, which was approved by the FDA in 2014 for use in relapsed/refractory PTCL based on results of a single-arm 129 patient trial that, to date, has only been published in abstract form.12 The patient population was typical of this group with the exception that prior treatment with a HDAC inhibitor was not permitted. The ORR was 26% with a CR rate of 11% and a DOR of 8.4 months. The grade 3-4 toxicity >10% was anemia 11% demonstrating that it was extremely well tolerated.
Available drugs/combinations
Bendamustine, reported in 60 patients, has a 50% ORR and 28% CR but the DOR of all patients is only 3.5 months.14 Gemcitabine, reported in 20 patients, has an ORR of 55% with 30% CR with a median PFS in the 6 responding patients of 34 months.15 The National Comprehensive Cancer Network (NCCN) guidelines list 6 combinations [DHAP (dexamethasone, cisplatin, cytarabine), ESHAP (etoposide, methylprednisolone, cytarabine, cisplatin), EPOCH (etoposide, vincristine, doxorubicin, cyclophosphamide, prednisone), GDP (gemcitabine, dexamethasone, cisplatin), GemOx (gemcitabine, oxaliplatin), ICE (ifosfamide, carboplatin, etoposide)], but there are only data for modified ESHAP,21 Gemcitabine-based22,23 regimens and ICE24 in PTCL. The data in Table 3 support the fact that although response rates are high, the duration of response is short even in less heavily pretreated patients who had a median of only 1 prior therapy, which demonstrates that for most patients even second-line therapies are not providing cures.
Role of transplant in the relapsed setting
In the relapsed setting there is general consensus that stem cell transplant should be used in eligible patients. Prospective clinical trial results are not available to inform us about the role of allogeneic versus autologous stem cell transplant at the time of disease relapse. However, there are 6 retrospective reports available all of which demonstrated inferior survival in the patients who received allogeneic stem cell transplant.25-30 The largest single institution report included 76 patients with T-cell lymphoma who underwent transplant for relapsed disease at MD Anderson between 1990 and 2009.27 Forty-one patients with a median age of 56 years, median of 2 prior therapies (76% of whom had chemosensitive disease) underwent autologous stem cell transplant (ASCT). Thirty-nine percent had PTCL-NOS, 34% had ALK- ALCL, 12% with angioimmunoblastic T-cell lymphoma, and 12% had extranodal t cell lymphoma. The 35 patients who received an allogeneic stem cell transplant (AlloSCT) were younger with a median age of 35 years but were more heavily pretreated with a median of 3 prior therapies and only 51% had chemosensitive disease at the time of transplant. In addition, 43% of them had extra-nodal T-cell lymphoma, predominantly natural killer T-cell lymphoma (26%), and only 11% had alk- negative ALCL. There was no difference in 4 year PFS (38% ASCT vs 28% allo SCT), but the 4 year OS of the patients undergoing autologous SCT was significantly better at 50% versus only 36% in the allogeneic group (p < 0.05).27 The inferior OS survival seen with alloSCT was in large part due to increased non-relapse mortality (NRM), with a 3 year NRM as high as 39% in 1 report versus 14% with ASCT (p = 0.024).26
The Center for International Blood and Marrow Transplant Research (CIBMTR) is a working group of > 500 transplantation centers worldwide which reviewed their registry data showing 115 autologous transplants and 126 allogeneic transplants for ALCL, PTCL, NOS, and AITL.30 Noting that there were more poor prognostic factors in the allogeneic transplant group such as transplant beyond CR1, chemotherapy refractory disease, non ALCL histology, and three or more chemotherapy regimens before transplant, they accounted for these differences in a multivariate analysis of prognostic factors for autoSCT and alloSCT recipients. The non-relapse mortality relative risk was 1.0030 for autoSCT and 3.543 for alloSCT, p < .001; the respective relapse progression was 1.0030 v = 0.728, p < .1738; and the overall mortality was 1.0030 v = 1.425, p = .0883. Although it is not surprising that the non-relapse mortality is significantly higher, it is a surprise that there is no difference in relapse rates. Based on these data, the traditional advantage of allogeneic transplant in controlling disease is not present in sufficient amount to compensate for the increased non-relapse mortality resulting in an overall mortality favoring autologous transplant. Based on the cumulative data, autologous stem cell transplant is the more frequently used approach in the relapsed setting.
Retrospective studies have consistently reported significantly improved OS in patients with chemosensitive disease at that time of transplant and the newly approved agents for PTCL (pralatrexate, romidepsin, belinostat, brentuxiab vedotin) will enable additional patients to achieve that goal. However, even for patients with chemo-refractory disease transplant should not be completely excluded as 3 year OS as high as 36% have been reported.25,26
Single agents in development
One of the most interesting drugs under investigation for use in ALCL is the ALK inhibitor, crizotinib. Crizotinib is FDA approved for use in patients with ALK+ non-small cell lung cancer. Early data suggests that ALK inhibitors will also be effective in the ALK+ lymphomas. A phase I study of crizotinib in pediatric patients included 9 patients with rel/ref ALK+ ALCL.31 A CR occurred in 7/9 patients (78%), 1 patient had a PR and the 9th patient had stable disease for an ORR of 88%. A similar ORR of 90.9% has been reported in 11 patients with rel/ref ALK+ ALCL who were treated as part of a compassionate use protocol of crizotinib.17 At 2 years the OS was 72.7% and the PFS was 63.7% with 1 patient on drug for >40 months and still in a remission. ALK inhibitors are not currently being used to treat ALK+ ALCL outside of the research setting, but clinical trials are underway in this patient population.
Mogamulizumab is a defucosylated humanized anti CC chemokine receptor 4 (CCR4) antibody. Chemokines are involved in the control of leukocyte migration and CCR4, which is expressed on 30%-78% of T-cell lymphomas, has been associated with inferior outcomes.32-34 A phase II study of mogamulizumab in rel/ref CCR4 + PTCL (n = 29) and cutaneous T-cell lymphoma (CTCL) (n = 8) reported an ORR of 34% and a CR/CRu rate of 17% in the patients with PTCL.34 However, a phase II trial performed in Europe had less encouraging results with an ORR of only 11% in 35 patients with rel/ref PTCL.35 At this time it is not clear whether active research for rel/ref PTCL will continue.
Alisertib is a reversible inhibitor of aurora A kinase, inhibition of which can lead to cell cycle arrest by affecting centrosome separation in mitosis.36 A phase II trial of alisertib in rel/ref aggressive non-Hodgkin lymphoma (n = 48) reported an ORR of 50% in the 8 patients with PTCL.37 However another phase II trial of alisertib in 37 patients with rel/ref PTCL and transformed mycosis fungoides (SWOG 1108)38 showed less promising results with an ORR of 24%, although it was 30% (CR 6.7%) when only the PTCL patients (n = 30) were considered. Results of a randomized phase III clinical trial of alisertib versus physician's choice of gemcitabine, pralatrexate or romidepsin are awaited with interest (NCT01466881).
Duviliseb is an oral inhibitor of phosphoinositide-3-kinases (PI3K-δ and PI3K-γ). These PI3Ks are involved in cell signaling with a role in growth of malignant T cells. A clinical trial of duviliseb in 33 patients with rel/ref T-cell lymphomas (16 PTCL and 17 CTCL) reported an ORR of 47% and CR of 13% in PTCL39 demonstrating that this might be another interesting pathway to target in the treatment of these difficult diseases.
Immune check point inhibitors such as antibodies that block programmed death-1 (PD-1)/programmed death ligand (PD-L1) and cytotoxic T lymphocyte associated protein 4 (CTLA-4) pathways are of interest in lymphomas because of their ability to reverse local T-cell immune tolerance potentially leading to increased antitumor response. The first of these agents, ipilimumab, a monoclonal antibody that blocks CTLA-4 was FDA approved for use in melanoma in 2011. Research into the use of immune checkpoint inhibitors in lymphoma, in particular into T-cell lymphomas, is in its infancy. At this time, there is no clinical data available about the efficacy of these agents in PTCL; however, pembrolizumab, an anti-PD-1 monoclonal antibody, is currently being studied in patients with rel/ref mycosis fungoides and sezary syndrome (NCT02243579). In the coming years clinical trials of immune check point inhibitors in PTCL will demonstrate its role, if any, in this difficult to treat disease. Even with increasing treatment options for PTCL, with the exception of ALCL, the outcomes for this patient population remain poor and therapeutic advances are desperately needed.
Combinations in development
In an effort to find the cure for PTCL, many clinical trials are underway exploring novel multidrug chemotherapy combinations as well as new drug targets. However, so far we have not found another highly effective drug, like brentuximab vedotin, so much more work is needed to improve outcomes for this patient population.
Since the approval of pralatrexate, romidepsin, belinostat, and brentuximab vedotin, there have been a flurry of studies combining these drugs with a CHOP backbone in hopes that they would have high efficacy in the front line setting.
Brentuximab vedotin is farthest along in this assessment as there is an ongoing randomized control trial (No. NCT01777152) of CHOP versus BV-CHP (brentuximab vedotin, cyclophosphamide, doxorubicin, prednisone) in patients with newly diagnosed CD30-positive mature T-cell lymphomas. A phase I trial of sequential BV followed by CHOP (n = 13) or concurrent BV-CHP (n = 26) had encouraging results.40 The ORR/CR rates were 85%/62% for the sequential group and 100%/88% for concurrent treatment. These are exciting responses but results of the randomized control trial (RCT) are required before it is routinely incorporated into the front-line setting.
The HDAC-I, romidepsin, has also been combined with CHOP in a 37 patient phase I/II trial.41 In 35 evaluable patients the CR/PR rates were 51%/17% with an estimated 18 month PFS of 57% and OS of 76.5%. Three of 37 patients had a cardiac event (myocardial infarction in 2 and acute cardiac failure in 1); although the connection to romidepsin is not clear, this will need close monitoring as studies move forward. This combination is being further explored in an ongoing phase III RCT (NCT01796002).
Pralatrexate alternating with CEOP (cyclophosphamide, etoposide, vincristine, prednisone) was also studied in patients with newly diagnosed PTCL.42 Unfortunately, the ORR of 70% and CR rate of 45% were not higher than CR rates in historical controls so this particular regimen is not an area of ongoing research, but a pralatrexate + concurrent CHOP protocol is in development.
It is possible that one of these multidrug regimens will improve outcomes for patients with PTCL, but there will still be a large number of patients with relapsed/refractory disease for whom new approaches are needed and there are several drugs under investigation that target new pathways of interest.
Conclusion
Progress is being made in the management of PTCL, but with the exception of Alk+ ALCL, the prognosis continues to be poor. Moving forward many questions still need to be answered, such as whether new drug combinations with the CHOP backbone or alternative chemotherapy regimens will improve survival, the utility of stem cell transplant in first remission, and ultimately the development of new targeted therapies that can improve chances of long-term survival in this difficult group of diseases.
As our understanding of the PTCLs increases we may be able to more effectively target different subtypes leading to improved outcomes. The recently developed novel anti-CD30 drug antibody conjugate, brentuximab vedotin, is the first drug to successfully employ this approach for the treatment of anaplastic large cell lymphoma (ALCL) which will lead to a drastic management change. Unfortunately, similar advances have not been made for the other PTCLs, and until they are, clinical improvements for this group are likely to be minimal.
How I treat PTCL
Many questions remain with regards to optimal management of PTCL; for this reason, when available, a clinical trial is always my first choice for treatment of this difficult disease. Outside of a clinical trial, first line therapy is typically CHOP or an etoposide-containing regimen, such as EPOCH or CHOEP with consideration of stem cell transplantation in first remission.
At the time of relapse of PTCL, NOS, a wide variety of options are available and there is no data clearly demonstrating that any agent or multiagent drug combination is superior to the others. My approach for patients who are transplant candidates and are symptomatic from their disease is typically a multidrug combination, often a gemcitabine-containing regimen rather than ICE, because most have already received etoposide. If they have chemosensitive disease then I will send them for autologous stem cell transplant.
For patients who are not transplant candidates or who have primary refractory PTCL NOS, I will frequently use one of the new agents, such as romidepsin, belinostat or pralatrexate, in hopes that new drug mechanisms will be effective in patients with chemo-refractory disease. For relapsed ALCL, my first-line approach is brentuximab vedotin. If this is not effective, then I follow the same track as outlined above for PTCL, NOS.
Unfortunately, even when receiving optimal care, with the exception of ALK+ ALCL, the majority of patients will not be cured of their T-cell lymphoma. However, there is a small group of patients who will obtain a long-term remission and so it is important to treat each new patient aggressively in hopes that they will be one of the fortunate ones.
Correspondence
Anne Beaven, Duke University Medical Center, Box 3872, DUMC, Durham, NC 27710; Phone: 919-684-8964; e-mail: Anne.beaven@duke.edu.
