
Abstract
The diffuse aggressive large B-cell lymphomas are a heterogeneous group of B-cell malignancies. Although many are readily recognized due to characteristic clinical and pathologic features, several problematic areas still exist in diagnosis of these lymphomas due to a variety of reasons that include imprecise or difficult-to-apply diagnostic criteria, gaps in our understanding of lymphoma biology, and limitations in technologies available in the clinical laboratory compared to the research laboratory. This may result in some degree of confusion in the pathology report, particularly if the issues are not clearly explained, leading to frustration or misinterpretation on the part of the reader. In this review, I will discuss the pathologic features of a subset of the WHO 2008 classification diffuse aggressive large B-cell lymphomas, focusing on areas in which difficulties exist in diagnosis and/or biomarker marker assessment. A deeper understanding of the issues and areas of uncertainty due to limitations in our knowledge about the biology of these diseases should lead to better communication between pathologists and clinicians.
Learning Objectives
Recognize clinical and pathologic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas
Be familiar with the concept of EBV-positive B-cell lymphomas in non-immunocompromised patients
Understand the concepts of cell of origin in diffuse large B-cell lymphoma and differentiate between the double-hit and double-expresser lymphomas
The diffuse aggressive large B-cell lymphomas are a heterogeneous group of B-cell malignancies. Recognized entities in the WHO 2008 classification are shown in Table 1.1 Distinctive entities with characteristic and specific pathologic and/or clinical features have been recognized, and include entities, such as plasmablastic lymphoma, primary effusion lymphoma, lymphomatoid granulomatosis, and intravascular lymphoma. Application of relatively unambiguous diagnostic criteria in combination with clinical presentations typically allow specific diagnoses. However, several problematic areas exist due to a variety of reasons that include imprecise or difficult-to-apply diagnostic criteria, gaps in our understanding of lymphoma biology, and limitations in technologies available in the clinical laboratory compared to the research laboratory (in which entities or biomarkers were defined). This may result in some degree of confusion in the pathology report, particularly if the issues are not clearly explained. In this review, I will discuss the pathologic features of a subset of the WHO 2008 classification diffuse aggressive large B-cell lymphomas, focusing on areas in which difficulties exist in diagnosis and/or biomarker marker assessment. In particular, I will focus on primary mediastinal large B-cell lymphoma, B-cell lymphoma, unclassifiable with features intermediate between diffuse large B-cell lymphoma and Hodgkin lymphoma, EBV+ diffuse large B-cell lymphoma of the elderly, diffuse large B-cell lymphoma not otherwise specified (DLBCL, nos), and B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma.

Primary mediastinal large B-cell lymphoma (PMBL)
PMBL is an uncommon lymphoma, accounting for 2.4% of NHLs.2 It typically occurs in young adults (median age 32 years) with a female predominance and presents as a bulky mediastinal mass that can invade local structures including lung, pleura, and pericardium. Spread to regional lymph nodes including cervical and supraclavicular nodes may be seen. Thus, most patients (75%) present with stage I or II disease. Because of the mass effect, patients may suffer from superior vena cava syndrome. Other common features include presence of pleural effusion and B-symptoms. Although spread to distant sites may occur, involvement of the bone marrow is very uncommon.3-5 The putative cell of origin is the thymic B-cell and gene expression profiling studies have demonstrated a PMBL gene signature, supporting the consideration of this lymphoma as a distinct entity.6,7
Histologically, the pattern is diffuse but this lymphoma may have a varied appearance. The lymphoma is composed of medium-to-large sized cells with round to lobulated nuclei, some of which may resemble Hodgkin cells. The cells often have moderate to abundant amounts of pale cytoplasm. There is frequently a delicate, compartmentalizing fibrosis. The immunophenotype is that of a mature B-cell expressing PAX5, CD19, CD20, and CD79a. CD10, BCL6, and MUM1 are variably expressed and CD21 is absent. BCL2 is often expressed (80%).8-10 CD23, a marker expressed on normal thymic B cells, is expressed in up to 70% of PMBL cases.11 CD30 is also commonly expressed but is generally weak and heterogeneous in pattern.12 Although OCT2, BOB.1 and PU.1 are consistently expressed, PMBL cases often lack detectable immunoglobulin by flow cytometry.10 Unfortunately none of these markers are specific for PMBL and may be expressed in DLBCL, nos. MAL, a gene involved in lipid raft organization, apical transport in epithelial cells, and T-cell signaling is also overexpressed in PMBL and immunohistochemical staining shows expression in 54%-70% of cases, whereas <5% of diffuse large B-cell lymphomas express MAL protein.13-15 EBV is only rarely, if ever, present in PMBL.16 BCL2, BCL6, and MYC rearrangements are not features of PMBL.17-19
Gene expression profiling studies not only demonstrated that PMBL was distinct from DLBCL nos but also that the profile and deregulated pathways showed similarities to classical Hodgkin lymphoma (cHL).7,17 This has implications both in diagnosis and overlap cases to be discussed shortly.
Genetic studies have demonstrated recurrent structural genetic changes to include gains of chromosome 9p24 in up to 79% of cases (JAK2, PDL1, and PDL2), 2p in ∼50% of cases (REL and BCL11A), Xp11.4-21 and Xq24-26 in ∼30%.20-22 Other studies have also implicated copy number alterations or mutations in JMJD2C, SOCS1, BCL11A, STAT6, TNFAIP3, MYC, and TP53, or epigenetic changes (promoter hypermethylation) in CDKN2A.18,23-29 These alterations can be generally organized into specific pathways related NFκB activation, JAK/STAT signaling, transcriptional regulation, cell cycle/TP53 pathway, and histone modification.30 More recently, recurrent translocations involving CIITA, a master regulator of HLA class II expression regulation, have been discovered in 38% of PMBL cases, and help explain decreased HLA class II expression seen in PMBL.31 Thus, avoiding immune surveillance may be a theme in PMBL. Indeed PDL1 and PDL2 have been identified as partner genes in CIITA translocations, the end result of which is to overexpress these PD1 receptor ligands under the control of the CIITA promoter.30,31 This is not the only mechanism for increased expression PDL1 and PDL2 because the above mentioned gains in chromosome 9p have been shown to result in increased expression of these molecules.23,32,33
Although much as been learned about the pathobiology of PMBL, application to everyday diagnostic practice has lagged. Assessment for gene expression signatures or rearrangements of CIITA, PDL1, and PDL2 have not made their way to the clinical laboratory due to factors including technological limitations or lack of commercially available reagents. Furthermore, although rearrangements in CIITA, PDL1, and PDL2 appear to be relatively specific for PMBL, sensitivity is modest. Thus, a major diagnostic dilemma faced by pathologists is distinguishing PMBL from DLBCL, nos presenting with a mediastinal mass. Pathologists still rely on classic PMBL pathologic findings of a diffuse infiltrate of large, often multilobulated, cells with variable amounts of pale cytoplasm, set in a fine compartmentalizing fibrotic stroma. Immunophenotypic clues favoring PMBL include variable expression of CD30 and CD23 and these markers, in association with other mature B-cell markers, and exclusion of other specific types of diffuse aggressive B-cell lymphomas (such as Burkitt lymphoma, lymphoblastic lymphoma, or transformed follicular lymphoma), and classical Hodgkin lymphoma (to be discussed in the next section) should allow a specific and definitive diagnosis in many cases when considering the constellation of clinical and pathologic features. To further complicate the issue, cases of diffuse large B-cell lymphoma have been identified that have the gene expression signature of PMBL despite the fact that there is no detectable mediastinal mass.34 Four of 6 such cases had pathologic features of PMBL such as fine sclerosis or variable expression of CD30 and CD23. However, one was felt to histologically be a DLBCL, whereas the other case had gray zone features between cHL and PMBL.34 Thus, one may now consider the possibility of a “non-mediastinal PMBL.”
Newer markers are now being examined and may further enhance one's ability to specifically and positively identify PMBL from other diffuse aggressive large B-cell lymphoma, notably DLBCL, nos presenting with a mediastinal mass. Among the proposed useful markers are expression of MAL, PDL2, and CD200, and coexpression of cytoplasmic TRAF1 and nuclear cREL.14,35-37 Given the current lack of suitable commercially available MAL and PDL2 antibodies and difficulties in cREL staining (as well as reported modest diagnostic sensitivity of 53% for cREL and TRAF1 coexpression), CD200 may be more promising but requires verification in other centers. Of note, we are currently evaluating the performance of a commercially available MAL antibody for use in identifying PMBL. Due to the lack of highly sensitive and specific phenotypic markers, pathology reports may reflect a degree of diagnostic uncertainty in some cases of suspected PMBL. Close communication between pathologists and clinicians may be required to agree on the most appropriate diagnosis.
B-cell lymphoma, unclassifiable with features intermediate between DLBCL and classical Hodgkin lymphoma
Because PMBL and cHL share biologic features at the molecular level, it stands to reason that some degree of diagnostic overlap might exist in which pathologic features are intermediate between the two entities. Indeed cases with sequential NHL and cHL, composite lymphomas, and cases with features of both a DLBCL and cHL have long been recognized.38-40 The WHO 2008 classification recognized the existence of cases that might have pathologic features intermediate between these two entities, commonly referred to as mediastinal gray zone lymphoma (MGZL).1 These cases, occur in younger patients with a median age of 20-40 years and a male predominance. Initial descriptions recognized 2 groups of cases.41 The first are those in which the initial impression is that of PMBL. However, occasional RS-like cells or nodules may be present. Additionally, phenotypic features may be atypical such as weak/absent CD20, expression of CD15, and weak expression of B-cell transcription factors such as PAX5, OCT2, and BOB.1, which are reminiscent of Reed–Sternberg cells. Alternatively, other cases resemble nodular sclerosis Hodgkin lymphoma but have features, such as strong CD20 expression, expression of CD79a and/or CD45, lack of the characteristic mixed inflammatory background, lack of necrosis, and have increased large mononuclear Hodgkin-like cells that are more in keeping with PMBL.41,42 Thus, there is a histologic spectrum of cytologic appearances in the same case, which can also vary in different areas of the tumor, and mixed immunophenotypic signals.
This theme of overlap also extends to molecular features since common genetic abnormalities such as gains in chromosome 2p, 9p, 16p, and 8q are found in both cHL and PMBL. Interestingly abnormalities in these same loci have been found in MGZLs.43 FISH studies showed gains or amplifications in 2p16.1 (BCL11/REL) in 33% of cases, gains of 8q24 (MYC) in 27%, and gains or amplifications of 9p24.1 (JAK2/PDL2). CIITA probes showed gain or amplification in 10% and translocation in 27% of cases.43 Of note, the frequency of translocation is intermediate between the 38% described for PMBL and 15% for cHL.31 Given the fact that metachronous cases of PMBL and cHL have occurred, suggesting some degree of “reprogramming” of the cells, one could surmise that epigenetic mechanisms might be relevant in this process. Methylation profiling of PMBL, cHL, and MGZL cases has shown distinct profiles for each type and a 22-gene methylation class prediction model was developed. Interestingly, composite PMBL/cHL cases (even when microdissected and individual components analyzed separately) maintained the profile of MGZL rather than splitting into PMBL and cHL components.44 In aggregate, these data appear to provide evidence and rationale for the relatedness of these lymphomas and support for identifying MGZL as a separable entity within the group.
As can now be understood, pathologists and clinicians are faced with the issue of imprecise diagnostic criteria for this overlap entity. In practice, pathologists should reserve the diagnosis of MGZL for cases that truly send mixed/hybrid signals; namely, morphology suggested one entity (PMBL or cHL) but phenotypic data more in keeping with the other, ideally supported by more than one aberrant marker. For example, cHL morphology with strong expression of CD20 and robust coexpression of B-cell associated transcription factors OCT-2 and BOB.1 or cases with PMBL morphology with expression of CD15 and downregulated OCT-2 or BOB.1. Likewise, minor morphologic variation, such as the occasional RS-like cell in typical PMBL, should not be the sole reason for designating a case as MGZL. Clearly stating the data supporting the intermediate features in the report will help the clinician understand the reasoning for arriving at the diagnosis. Recognizing that composite cases can occur, MGZL designation should not be used for such composite lymphoma cases.42 Of note, the methylation profiling data might suggest that these composite cases, from a clinical perspective, could be managed as a MGZL case. Finally, one should avoid a definitive diagnosis of MGZL in a needle biopsy given the inherent morphologic spectrum of these cases and need to exclude composite lymphomas in this setting.
EBV-positive DLBCL of the elderly
EBV-positive DLBCL of the elderly as defined in the WHO 2008 occurs in adults over the age of 50 who have no known immunodeficiency or other specific EBV+ lymphoproliferative disorder, such as lymphomatoid granulomatosis or EBV+ DLBCL associated with chronic inflammation. It is believed to be related to deterioration of the immune system that may occur in elderly patients (immune senescence). It comprises 8%-10% of DLBCL in Asian countries but is likely lower (3%) in the United States.45-47 Most patients (80%) present with lymphadenopathy. Extranodal involvement may occur and often involves skin, lung, tonsil, and stomach. Elevated LDH (58%) and B symptoms (49%) are common.48 This lymphoma is aggressive and has a poor prognosis compared to EBV-negative tumors and prognostic factors include presence of B symptoms and age >70 years.46
Pathologically, cases show architectural effacement by an atypical large cell infiltrate. The cells often show a morphologic heterogeneity, with a polymorphous infiltrate demonstrating varying degrees of plasmacytic differentiation. Large centroblasts, immunoblasts, and multilobated Reed–Sternberg-like cells can be seen. Necrosis is also a characteristic feature. Immunophenotyping shows expression of CD20 and other pan-B cell markers such as CD79a. A non-germinal center B-cell phenotype with MUM1 expression is often seen and reflects the plasmacytic maturation.45,46 By definition EBV (best assessed by EBER staining) is positive in the vast majority of cells. CD30 is expressed in a high proportion of cases (75%).46 RS-like cells may express CD15 and show decreased CD20 expression, but usually retain expression of PAX5 and OCT2, unlike true RS-cells in which OCT2 is downregulated. Thus, classical Hodgkin lymphoma may become a consideration. Features that favor an age-related EBV-positive lymphoproliferative disorders/lymphoma include extranodal involvement, lack of the typical mixed inflammatory background of Hodgkin lymphoma, and the variation in cell morphology expressing CD20, CD30, and EBER.
Other EBV+ lymphoproliferative disorders (expanding the spectrum of disease)
The spectrum of EBV-positive lymphoproliferative disorders has been expanded to include reactive and neoplastic processes, described are reactive lymphoid hyperplasia, polymorphic extranodal and nodal LPDs, and DLBCL. Polymorphic extranodal LPDs were divided into isolated lesions and lesions that fit descriptions of EBV+ mucocutaneous ulcer.49 This latter lesion is a relatively recently described entity in which an EBV+ superficial ulcerating lesion involving gastrointestinal tract, skin, and oropharyngeal mucosa occurs. It is composed of a polymorphous infiltrate with a spectrum of cells to include atypical large B-blasts and Reed–Sternberg-like cells. Despite the cytologically atypical appearance, prognosis is excellent with conservative therapy.49,50 Although the polymorphic extranodal LPDs had a good prognosis (5 year disease-specific survival of 93%), the nodal polymorphic LPDs and DLBCL cases had poor outcomes (5 year disease-specific survival of 57% and 25%).49 Distinguishing features for DLBCL include lack of the full spectrum of plasmacytic differentiation, less variation in cell size, and marked pleomorphism compared with nodal polymorphic LPDs.
Immunoglobulin heavy chain gene rearrangements are seen more frequently in EBV-positive polymorphic nodal lymphoproliferative disorders and DLBCL categories (56%-63%) compared to reactive hyperplasia and extranodal lymphoproliferative disorders (16%-33%). Interestingly T-cell receptor gamma gene rearrangements were detected in 27%-50% of reactive hyperplasia and extranodal lymphoproliferative disorders and 15%-24% of polymorphic nodal lymphoproliferative disorders and DLBCL cases.49 The presence of T-cell clones is felt to reflect the limited T-cell repertoire in the immunesenescent setting.51-54
It has been recognized that the somewhat arbitrary age limitation of 50 years may not be appropriate. A series of 46 cases was reported in patients <45 years of age. The median age was 23 years with a male–female ratio of 3.6. All patients presented with lymphadenopathy and only 11% had extranodal disease. Interestingly the histopathology was most commonly that of T-cell histiocyte-rich large B-cell lymphoma (78%), mediastinal gray zone lymphoma (15%), and DLBCL (7%). Tumor cells often expressed CD30, as well as PDL1 and IDO, the latter markers suggesting a local immunosuppressive or immune tolerant microenvironment.55,56
This entity has altered practice and increased the necessary work-up in large B-cell lymphomas. Very little data exists on how frequently EBV is assessed by pathologists in cases of diffuse aggressive B-cell lymphoma. However, given the prognostic implications, assessment should be routinely performed. Fortunately, there are known histopathologic and phenotype clues that can focus efforts at EBV detection. These include a polymorphous infiltrate with plasmacytic differentiation, presence of Hodgkin-like cells, necrosis, and non-germinal center B-cell phenotype. Understanding the spectrum of EBV-positive LPDs is important so that hyperplastic processes and extranodal polymorphic lesions, particularly EBV-positive mucocutaneous ulcer are not overinterpreted as overt lymphoma.49 On the other hand, understanding that EBV-positive DLBCL in the older adult population and polymorphic LPDs in lymph nodes that resemble polymorphic post-transplant lymphomas are aggressive B-cell lymphomas will prompt consideration of appropriate conventional immunochemotherapy and, perhaps in the future, strategies to improve immunity toward EBV. Recognizing advances in our understanding of EBV-driven B-cell LPDs may affect nomenclature in the anticipated WHO update. Cases of EBV+ DLBCL of the elderly may be designated as EBV+ DLBCL nos, without reference to age of the patient.
Diffuse large B-cell lymphoma, not otherwise specified: biomarkers and risk assessment
The diagnosis of DLBCL, nos is generally straight forward, but currently there is a degree of uncertainty with regard to the types of studies for risk assessment that should be done and what techniques to use. Pathologist's tasks are twofold when making a diagnosis of DLBCL, nos. The first is to exclude other large B-cell lymphoma entities and subtypes of DLBCL (Table 1), whereas the second is to perform biomarker assessment that might yield important prognostic or risk assessment information. The pathology report should contain enough relevant information for both these purposes and we will concern ourselves with these two issues.
Other forms of diffuse aggressive B-cell lymphomas, such as PMBL, intravascular lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, ALK+ large B-cell lymphoma, oral plasmablastic lymphoma, and HHV8-related lymphomas (primary effusion lymphoma and lymphomas occurring in the setting of multicentric Castleman disease) are usually distinguishable from DLBCL nos by a constellation of characteristic clinical and pathologic features. Because of the great importance of clinical setting on these diagnoses, pathologists must actively attempt to understand the clinical setting and clinicians should make every attempt to inform their pathologists about relevant clinical information, such as location/distribution of disease, HIV status, or other potential causes of immunocompromise. In addition, pathologists must use sufficient ancillary studies to exclude these subtypes and other forms of lymphoma. Discretion of course can be used, with certain markers done secondarily when other primary markers suggest a particular lymphoma type is more likely to be in the differential diagnosis based on the clinical, histologic, and phenotypic features. For example, ALK does not need to be routinely assessed unless the proper morphology (immunoblastic) and plasmablastic phenotype are present. Likewise, other entities that may be confused with DLBCL, nos should be excluded, such as pleomorphic blastoid mantle cell lymphoma. Table 2 shows an example diagnostic panel and conditions in which markers might be secondarily assessed. Use of such a panel with described results in the body of the report should allow the pathologist to convey the differential diagnostic considerations of the case and the supporting evidence for the final diagnosis.
Example diffuse aggressive B-cell lymphoma marker panel
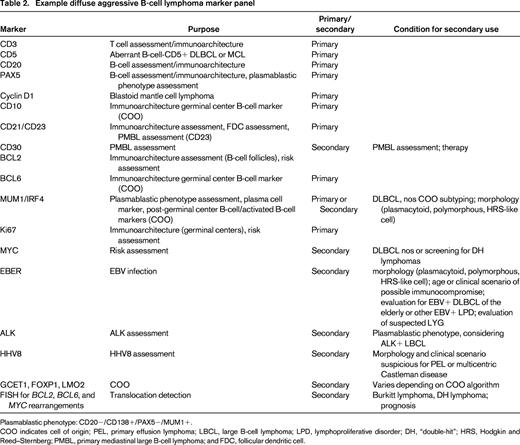
Plasmablastic phenotype: CD20−/CD138+/PAX5−/MUM1+.
COO indicates cell of origin; PEL, primary effusion lymphoma; LBCL, large B-cell lymphoma; LPD, lymphoproliferative disorder; DH, “double-hit”; HRS, Hodgkin and Reed–Sternberg; PMBL, primary mediastinal large B-cell lymphoma; and FDC, follicular dendritic cell.
One can see that the intensity of the work-up, even for something as seemingly straight forward as DLBCL, nos, is substantial and far greater than required in years past. Fortunately, the availability of these tests (whether done on-site or through reference laboratory services) in formalin-fixed paraffin-embedded tissues (FFPET) now enables any pathologist to have these studies performed. Unfortunately, we are still limited by the amount and quality of tissues provided by the biopsy. With the proliferation of guided needle core biopsies, the amount of tissue available for diagnosis is drastically decreased. In instances where architectural assessment is important, necrosis is present, or tissues are partially involved (common scenarios include mediastinal tumors and assessment of gray zone lymphomas, question of underlying follicular lymphoma, DLBCL versus grade 3 follicular lymphoma, EBV+ LPDs including mononucleosis), excisional biopsies may be required. However, many studies have suggested that needle core biopsy is a reasonable first option that can result in an actionable diagnosis in a majority of cases. The degree of acceptable result varies from study to study depending on whether an actionable result was defined as the outcome or whether definitive WHO subtype was required. Variability also derives from whether cases were limited to those in which lymphoma was present or a wider variety of diagnoses including reactive conditions were allowed. However, general themes are that close cooperation with hematopathologists, incorporation of ancillary testing, such as flow cytometry, immunohistochemistry, and molecular studies, as well as larger size and greater number of cores, optimize diagnostic yield.57-62 Fine-needle aspirate cytology should not be used for primary diagnosis of lymphoma.63
Biology and biomarkers in DLBCL, nos
Over the past 15 years, our understanding of the biology of DLBCL nos has improved and we now recognize the biologic heterogeneity of DLBCL nos that underlies and helps explain some of the clinical heterogeneity in terms of response and outcomes to modern therapies. Landmark gene expression profiling studies have defined several signatures that relate DLBCL nos to normal cell counterparts (cell of origin; COO).64-67 In particular, cases can be related to that of a germinal center B-cell (GCB) or an in vitro activated B-cell (ABC). In addition to elucidating some of the biological heterogeneity of DLBCL, nos, this dichotomization also has clinical relevance with the ABC type having a worse prognosis than GCB type, independent of known clinical risk factors such as the International Prognostic Index (IPI). These data have been confirmed in patients treated with rituximab-CHOP therapy.67 Other signatures were also defined with clinical relevance. A favorable stromal signature (stromal signature 1) was identified that reflects extracellular matrix deposition and tumor infiltrating macrophages. An unfavorable stromal signature (stromal signature 2) was also identified that reflects angiogenesis and high blood vessel density.67
In addition to these important expression signatures, genetic abnormalities have been discovered. For example, numerical abnormalities, such as trisomy 3 and deletion of CDKN2A, are associated with poor prognosis.68 In fact, these abnormalities track with the COO and further support the concept that different subtypes of DLBCL arise in different molecular genetic backgrounds, reflecting their biologic distinctiveness that is also manifest in clinical differences in outcome.68 Mutations in specific pathways that are associated with cell of origin have also been discovered. For example, mutations in the B-cell receptor signaling pathway leading to NFκB activation are associated with ABC type of DLBCL, nos and mutation in the EZH2 involved in histone modification is seen in GCB DLBCLs. Both pathways are potential targets of therapy.
High throughput sequencing studies have also begun to define the mutational landscape of DLBCL, nos.69-72 These studies have demonstrated sets of commonly mutated genes and suggested a number of important gene ontogeny categories. In one large study of 73 primary DLBLC cases 34 of which had paired normal DNA, 322 recurrently mutated candidate genes were identified. The genes could be organized into pathways and >50% of the genes could be placed into one of 12 gene ontologies. The most common processes were signal transduction (including genes in the JAK-STAT, ubiquitin, WNT, NFkB, NOTCH, and PI3 kinase pathways) and chromatin modification (including SETD2, MLL3, ARID1A, and MEF2B). The degree of non-overlap in gene lists found amongst studies likely reflects the high degree of biologic heterogeneity, with a long tail of uncommon but recurrently mutated genes.72 Table 3 shows selected commonly mutated genes and associated pathways from multiple next-generation-sequencing studies.
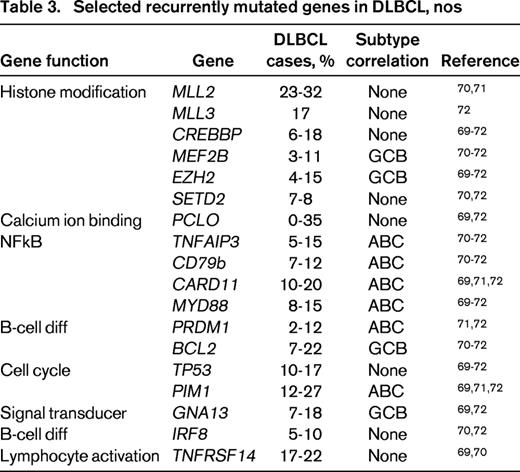
Despite the explosion of biologic data from broad scale gene expression profiling, array-based DNA copy number, and high throughput sequencing technologies, we have been challenged to reduce these discoveries to practice for reasons that include requirements for fresh frozen tissue, requirement for highly specialized equipment, potential long assay turnaround times, and cost. Clearly, for FFPET, immunohistochemistry (IHC) has great appeal for broad applicability and cost. However, issues related to standardization and pre-analytic variability that may affect antigen preservation and interobserver variability have been obstacles. Improvements in staining technology, advances in antibody technology, and experience in other tumor systems, such as breast cancer, with regard to standardization of tissue fixation, interpretive guidelines, and proficiency testing suggest that IHC biomarker assessment can be done, and but faces significant obstacles.73-77
In particular, there has been great interest in determining COO (GCB vs non-GCB) and IHC algorithms, generally using 3-5 markers have been developed.78-81 As can be seen from Figure 1, the algorithms differ in terms of approach, markers used, and cutoffs for each marker. Published concordance rates range from 86%-93%. Other performance characteristics such as sensitivity, specificity, and positive predictive value for GCB and ABC types also vary but range from 81%-99% (Table 4).78-81 The COO not only has prognostic value but is currently being used as an integral biomarker for clinical trials enriching for ABC types of DLBCL. Therefore, accurate determination and reporting of this data is becoming critical. In addition, based on the strength of the biological data and prognostic importance, it is likely the upcoming WHO Classification update will include COO determination as a subtype of DLBCL nos rather than a variant, requiring determination in daily practice.82

Cell of origin immunostaining algorithms and published performance
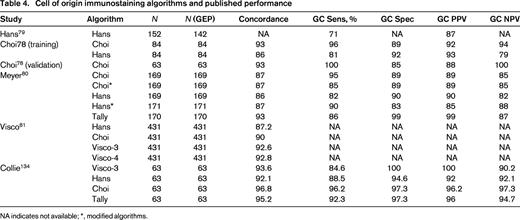
NA indicates not available; *, modified algorithms.
Advances in molecular technology have the potential to offer a molecular-based alternative to immunophenotypic surrogates. Experience has shown that whole genome expression profiling is not required to perform COO determination and limited gene sets have been identified. For example, Wright et al reported a 14 gene classifier capable of determining COO.66 This classifier has been reduced to practice using a single-tube multiplex capillary electrophoresis-based real-time QPCR assay and is currently available as a test in our clinical laboratories.83 A 20 gene classifier has also been reported and is being actively developed as a commercial test.84 Of note, such molecular assays may also be used to calibrate and validate IHC-based assays to maximize availability, minimize turnaround times, and decrease cost if adequate performance can be maintained. Indeed we have validated our IHC algorithms using such an approach with sensitivities and specificities for GCB and ABC subtypes in the 90% range (Tables 4 and 5).
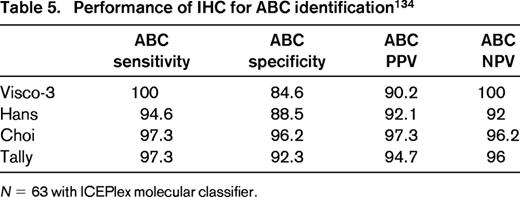
Can we use IHC in the routine laboratory for COO prediction? Given that molecular classifiers operate on a 90% probability threshold for determining molecular classification, IHC performance is arguably appropriate for use in the clinical setting, provided laboratories verify the IHC performance characteristics against gene-expression-based assays in their laboratories. In addition, the trend toward small biopsies or confounding issues such as presence of necrosis and inclusion of non-tumor tissues in biopsies may limit application of some molecular assays, thus IHC-based alternatives/back-ups may be required. With several possible IHC algorithms and now molecular test alternatives to choose from, pathology reports should be clear regarding which method is used and pathologists should adhere to the specified cutoffs if a particular IHC algorithm is used. Ideally, these should be reported in synoptic format for clarity. Reporting templates for biomarker data in clinical reports have prepared for this type of data and are published by the College of American Pathologists to serve as a guide for pathologists.85
In the search for biomarkers that assist in risk stratification, recent interest has turned to expression of MYC and BCL2 (double expressors; DEs). It is known that cases of DLBCL with MYC and BCL2 translocations have an extremely poor prognosis.86-88 However, other mechanisms besides translocations can result in overexpression of these proteins.68,89 With the development of a monoclonal antibody suitable for FFPET, 2 groups studied expression of BCL2 and MYC by immunohistochemistry.90,91 Green et al studied a series of 193 cases of DLBCL treated with R-CHOP and, using median expression values, determined cutoffs of ≥70% for BCL2 and ≥40% for MYC identified a high-risk patient population, which was then validated in a separate cohort of 116 patients.90 Johnson et al evaluated a training set of 167 DLBCL patients treated with RCHOP and determined cutoffs of ≥50% for BCL2 and ≥40% for MYC. This was validated in a separate set of 140 cases.91 Both series noted a strong association between DEs and COO classification (ABC type or non-GCB phenotype).90,91 Furthermore, the DE status was an independent predictor of PFS and OS when IPI and COO were included in statistical models. The adverse effect of DE held when genetic double-hit (DH) lymphomas were excluded.91
In a follow-up large multicenter study, Hu et al92 confirmed this effect by evaluating 40% and 70% cutoffs for MYC and BCL2 proteins, respectively, where cutoffs were arrived at through receiver-operator-curve analysis. The adverse impact on outcome was confirmed as well as the association with ABC type, independence from IPI, and was also shown once DH lymphomas were excluded. Of note, due to the high association with COO, ABC type was no longer associated with poor prognosis once DE status was included.92
Several other studies have been published examining DE status, each confirming the poor prognostic impact of DE status. However, cutoffs for BCL2 varied,90,91,93 and in 1 study treatment was not homogeneous.94 Thus, the preponderance of evidence shows the importance of DE status for risk stratification in DLBCL, nos; however, it should be mentioned that not all studies have shown the poor prognostic effects perhaps due to differences in study population.95 Notwithstanding, there are still some unsettled issues related to precise cutoffs to use in practice. Reproducibility has been shown to be good (91%) for BCL2.91 It also appears good for MYC, because one pathologist scored MYC in the training set of Johnson et al, and different pathologists scored the validation set.91 There is also general agreement on antibody clones for these markers because the Y69 clone for MYC and the 124 clone for BCL2 were used in all studies cited. We have recently validated these immunostains in our cohort of 69 RCHOP-treated DLBCL, nos patients, with the purpose of evaluating our technical platforms and the cutoffs of ≥40% for MYC and ≥70% for BCL2. Using automated immunostainers commercially available IVD kits for MYC (clone Y69) and BCL2 (clones 124 and SP66), we confirmed that ≥40%/≥70% cutoffs for MYC and BCL2, respectively, identified patients with poor OS, independent of the IPI. Furthermore, we found that ≥40%/≥50% also performed similarly and that clones SP66 and 124 also performed similarly.96 Thus, for laboratories wishing to implement this type of testing, these reagents appear suitable for use in this indication. Because a 50% cutoff for BCL2 may be easier to estimate, we suggest cutoffs of ≥40% for MYC and ≥50% for BCL2 be adopted, although those wishing to use 70% for BCL2 will be on firm footing. As with the COO, pathology reporting should be clear in terms of cutoffs used.
The microenvironment
Although much effort has been directed at understanding the biologic feature intrinsic to the tumor cells, features of related to immune response and microenvironment have been also been defined. As noted previously, gene profiling efforts identified stromal signatures that were of prognostic importance.67 Other gene expression studies have similarly found that microenvironmental components are differentially expressed and provide prognostic information.97-99 Attempts to reduce these findings to practice using immunohistochemical markers for gene products such as fibronectin and SPARC, microvessel density, or infiltrating T cells, Tregs, and macrophages have been successful in demonstrating feasibility, but have yet to be extensively reproduced to the same extent that COO or MYC/BCL2 expression have been.100-105 Interest is mounting in manipulating the immune microenvironment and reconstituting an anti-tumor response through PD1 blockade and a promising phase II trial has been completed in DLBCL.106 Further work, including whether assessment of PDL1 as a predictive biomarker is of value, is warranted.
B-cell lymphoma, unclassifiable with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma (BCLu) and “double-hit lymphomas”
BCLu
BCLu is an imprecisely defined entity in the 2008 WHO classification and was meant to recognize the overlap and gray zone between DLBCL and BL.1 Such cases were demonstrated in GEP studies, in which expression profiles intermediate between signatures of molecular BL (mBL) and DLBCL were seen.107,108 Histologic features vary. Some cases have high-grade features, such as starry sky and intermediately sized cells, but demonstrate excessive pleomorphism that exclude BL. Others may have morphologic features that closely resemble BL; however, phenotype (such as expression of BCL2) or genotype (concurrent MYC and BCL2 or BCL6 translocations, or MYC translocation with complex karyotype) make a case unacceptable for BL. Still others may have morphology of DLBCL but an intermediate GEP with or without MYC translocation.1,107,108 Too make matters worse, some case of mBL as defined by GEP, have genetic or morphologic feature that are atypical for BL, and include some cases with MYC and BCL2 translocations (“double-hit” cases) or complex genetic backgrounds with MYC translocations that would not be acceptable for BL. As a group, patients tend to be older and have a poor prognosis.109 Table 6 has the current 2008 WHO features of BCLu, with comparison to DLBCL and BL.
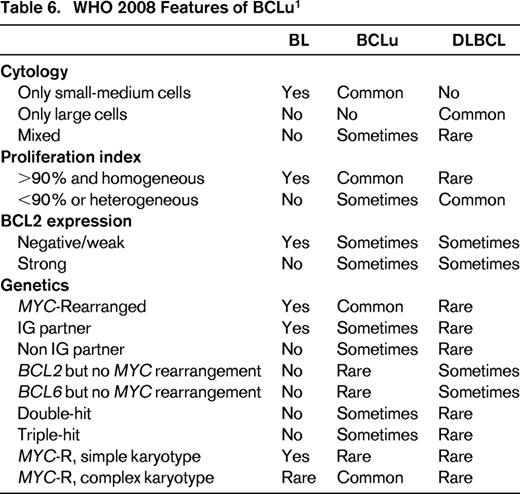
One can immediately see the problem in daily practice, because GEP and detailed genetic analysis by array-based and/or karyotyping is needed to recapitulate studies done to recognize BCLu. As a consequence, pathologists primary entry point for BCLu is appropriate morphology, ie, a case with “high grade” morphology (Table 6), which is then assessed for other supporting phenotypic or molecular features. An operational guideline would be to reserve this designation in a pathology report for cases that histologically closely resemble BL but in which the immunophenotype (such as lack of CD10, expression of strong BCL2) or genotype (complex karyotype, presence of BCL2 or BCL6 rearrangement along with MYC rearrangement, MYC rearrangement with non-IG partner gene) are unacceptable for BL. Some cases may have mixed morphology in which some intermediately sized Burkitt-type cells as well as large cells are present. As a corollary, cases with typical DLBCL, nos morphology, whether or not a MYC rearrangement is present, should not be placed into the BCLu designation. Also implied is that cases with appropriate morphology must be studied for genetic features that could be determined from routine karyotyping. Because karyotyping is not common practice, extensive FISH testing for MYC, BCL2, and BCL6 rearrangements becomes imperative (see below). How frequently does “high grade morphology” occur in diffuse aggressive B-cell lymphoma? In a review of diffuse aggressive B-cell lymphomas from a phase 3 trial of 260 patients, 12% were deemed to have BCLu morphology.110
Double-hit lymphoma
The term “double-hit” (DH) lymphoma, recently reviewed at the 2014 American Society of Hematology meeting education sessions, has been used variably in the literature and typically refers to cases with MYC translocation in combination of either BCL2 or BCL6 translocations.111 However, some studies have included CCND1 translocations or advocate inclusion of cases with numerical abnormalities in MYC or BCL2.87,112,113 Cases with CCND1 and MYC translocation are best considered as forms of mantle cell lymphoma (blastoid variants) and there is not consensus whether numerical abnormalities in MYC and BCL2 should be included. More data is needed to accept such cases in formal classification systems. DH lymphomas are thus not a specific entity in the WHO 2008. They represent approximately 2%-14% of DLBCL and 32%-78% of BCLu.111,113-116 Variation can be explained by different patient populations studied as well as differences in methods used to identify cases. The histopathology of DH or triple-hit (TH; rare cases with concurrent MYC, BCL2, and BCL6 rearrangements) lymphomas cases is variable. In select series where inclusion was largely based on molecular features, DLBCL nos and BCLu histology is reported in 32%-72% (median 62%) and 2%-62% (median 28%), respectively, with a small proportion (<15%) being follicular lymphoma or transformed from low grade lymphoma.86,88,112,113,117,118 Coming at the issue from the starting point of the histopathology, 2%-12% of DLBL are DH, whereas 32%-78% of BCLu are DH.111 Morphology may matter with some studies suggesting DLBCL, nos morphology has a better overall survival compared with non-DLBCL or BCLu morphology.86,88 The immunophenotype of DH lymphomas is heavily weighted to a germinal center B-cell phenotype with approximately 80%-90% of cases expressing CD10 and BCL6 with MUM1/IRF4 expressed in <20% of cases.86-88,119-122 In MYC/BCL6 DH cases, CD10 is less likely to be expressed compared with MYC/BCL2 DH cases.119 DH and the rare TH lymphomas generally have an aggressive clinical course with many studies showing mean or median survivals on the order of 1 year or less.86,88,114,117,118,123 Thus alternate treatments are needed.
Because routine karyotyping is seldom performed in NHLs, most cases are defined by fluorescent in situ hybridization (FISH) studies. FISH studies are most often used and depending on probes used, cases may not be recognized. In general, break-apart probes will be the most sensitive method to detect translocations; however, partner genes are not identified. Specific fusion probes are required to positively identify the fusion partners. For example, break apart probes for MYC will identify the great majority of translocated cases, but ∼10% of MYC translocated cases may be missed in break-apart only probes are used.123 However, IGH-MYC, IGK-MYC, and IGL-MYC probes must be used to determine whether the IG genes are involved in the translocation. Unfortunately, the latter 2 probe sets are not commercially available. If MYC rearrangement is present but IGH-MYC dual fusion probes are negative, break-apart IGK or IGL may be used to infer that these genes are involved; however, it is not definitive because it is possible that other genes may be involved in fusion with IGK or IGL.
Does the partner gene for MYC matter? Some studies,86,124,125 but not all,112 suggest that only IG-MYC fusions are associated with poor prognosis in DH lymphomas, whereas MYC fusion with non-IG partner genes is not associated with aggressive behavior.86,124,125 However, it should be remembered that most studies that have defined the very poor prognosis of DH lymphoma used break apart MYC probes to help identify cases.88,113,114,117,126-128 To further confound the issue of definition of DH, one study has suggested that so-called “atypical DH lymphomas” (those with single translocations of MYC or BCL2 with copy number alterations of the other gene, or copy number alterations of both genes) have similar poor outcome to conventional DH lymphomas.87,129,130 In addition to potential clinical importance associated with MYC partner genes, there may be histologic associations as well. A non-IG partner gene was associated with DLBCL morphology.86 The importance of MYC partner gene remains to be resolved.
In interest of full disclosure, not all studies have shown the importance of recognizing DH lymphomas. For example, Aukema et al in a series of non-uniformly treated patients found no difference in outcome between patients with SH and DH lymphomas.112 However, cases with a mBL gene expression signature were excluded and treatment protocols were predominantly in the pre-rituximab era. Tzankhov et al reported that in a multivariate analysis of DLBCL patients, MYC-R and high level of MYC protein expression were the most important factors along with IPI group and R-CHOP-like treatment in predicting outcome. Thus DH status lost its importance once these factors were known.123
In view of these data, it is not surprising that current laboratory practice is quite variable. Many laboratories test for MYC with break-apart probes. A subset may follow-up a positive result with IGH-MYC dual fusion probes. Very few laboratories if any, routinely attempt IGK and IGL FISH as secondary assays in the event that a MYC translocation is present while an IGH-MYC dual fusion probe set is negative. In an attempt to minimize cost, some laboratories have adopted screening using IHC for MYC. With the development of suitable MYC antibodies, one may enrich for translocated cases by only FISH testing cases that have MYC protein expression above a certain threshold, often 40%-50%.131,132 In a small series of 56 DLBCL, a threshold of 50% nuclear staining for MYC detected all cases with MYC translocation.132 However, results in other larger series has not been as good. With cutoffs ranging from 30% to 50%, the percentage of MYC-R cases below the cutoff ranged from 18% to 41% with lower cutoffs generally being associated with lower percentage of MYC-R DLBCL cases.90,93,95,123,128 Indeed, Horn et al argue against using a combined IHC/FISH algorithm in favor of FISH due to the potential for missing cases screened by IHC, but do concede that a 20% threshold could be considered (with a sensitivity of 92.3% for picking up MYC-R cases).95 One point should be made clear, Ki67 proliferative index should not be used as a screen because many cases of MYC translocated cases will have a low proliferative fraction. For example 42% of MYC translocated cases were shown to have a Ki67 proliferation index <80%.133 Practices seeking to maximize detection of DH lymphomas will likely adopt FISH testing in all diffuse aggressive B-cell lymphomas using MYC break-apart and IGH-MYC fusion probes along with BCL2 and BCL6 break apart probes. Those looking to use balance cost and sensitivity may adopt an IHC screen. However, individual laboratories should understand the performance characteristics of the MYC IHC stain and chosen cutoff for this purpose. Routine FISH testing will likely become obligatory as the much anticipated update to the current WHO classification may call for a high grade B-cell lymphoma, not otherwise specified (HGBCL, nos) parsed into 2 subgroups. The first is HGBCL,nos with MYC and BCL2 or BCL6 rearrangement (with specified morphology of either DLBCL, nos or BCLu). The second is HGBCL, nos without DH genetic features but with BCLu or other “high-grade” histologic features.82
Summary
The diagnosis, characterization, and reporting of diffuse aggressive large B-cell lymphomas has grown more complex. Molecular profiling has defined new entities, helped us recognize pathologic gray zones, revealed clinically relevant biologic subtypes, and helped identify prognostic and potentially predictive biomarkers. High-throughput sequencing studies are also providing insight into the mutational landscape that will inform new therapeutic strategies and perhaps provide a new framework with which to understand and classify these lymphomas. Inclusion of current relevant pathologic, immunophenotypic, and genetic features requires close interaction between pathologists and clinicians so that these data are presented in a clear and clinically meaningful way. Incorporation into local and national guidelines that are periodically reviewed and updated will drive the application in all types of practice settings.
Correspondence
Eric D. Hsi, Department of Laboratory Medicine, L30, Cleveland Clinic, 9500 Euclid Ave, Cleveland, OH 44195; Phone: 216-444-5230; Fax: 216-445-7253; e-mail: hsie@ccf.org.
