
Abstract
Myelofibrosis (MF) is the most aggressive form of Philadelphia chromosome–negative myeloproliferative neoplasm, and it is complicated by severe symptom burden, thrombotic events, infections, cytopenias, and transformation to acute myeloid leukemia (AML). Ruxolitinib, the first-line therapy for symptomatic or intermediate- and high–prognostic risk MF, has improved overall survival for this population. However, approximately one-half of MF patients will discontinue ruxolitinib by the first few years of therapy due to a spectrum of resistance, intolerance, relapse, or progression to blast phase disease. Danazol, erythropoietin-stimulating agents, and spleen-directed therapies can be useful in the ruxolitinib-resistant setting. In the ruxolitinib-refractory or -intolerant setting, commercial and novel therapies, either alone or in combination with ruxolitinib, have shown clinical utility. For blast-phase MF, the recent advancements in available AML therapies have increased the options with targeted and more tolerable therapies. In this article, we will discuss our paradigm for the management of relapsed/refractory and blast-phase MF in the context of therapeutic advancements in both AML and MF.
Learning Objectives
Understand the challenging clinical spectrum of ruxolitinib resistance, loss of response, and intolerance that has become increasingly common given the prolongation of survival with ruxolitinib use in myelofibrosis
Discuss optimization of ruxolitinib dosing using danazol, erythropoietin-stimulating agents, or spleen-directed therapies that can be helpful in the treatment of ruxolitinib resistance; for ruxolitinib-refractory or -intolerant patients, treatment can include clinical trials or commercially available therapies either as a single agent or in combination with ruxolitinib (ie, hypomethylating agents, lenalidomide, and histone deacetylase inhibitors)
Review that myeloproliferative neoplasm (MPN) blast-phase disease is associated with an aggressive course and very poor prognosis, with salvage chemotherapy and allogeneic stem cell transplant being the only curative treatment options
Apply individualized treatment approaches to blast-phase MPN patients who are not eligible for transplant, with a focus on disease features, clinical trial options, targeted therapy where appropriate with consideration of new available acute myeloid leukemia therapies, and prioritization of patient goals
Case 1
A 71-year-old male patient presents to your clinic with primary myelofibrosis (MF). He was diagnosed ∼10 years ago and is known to carry both calreticulin (CALR) and ASXL1 mutations. His Dynamic International Prognostic Scoring System Plus risk is Intermediate-2 (due to age >65 years old, hemoglobin of 7-8 g/dL, and the presence of constitutional symptoms). For the past 4 years, he has been maintained on ruxolitinib. His ruxolitinib dosing was initially titrated up to 20 mg twice daily, but due to worsening anemia, his ruxolitinib dose has been weaned down to 5 mg twice daily. Although he initially enjoyed a robust spleen and symptom response, over the last year, he has experienced progressive splenomegaly, fatigue, drenching night sweats, and abdominal discomfort that limit his quality of life. Despite having met with a transplant physician, he refused allogeneic transplantation due to personal concerns. He comes to you today to discuss options for treatment.
Introduction
MF is a type of myeloproliferative neoplasm (MPN) that originates from dysregulated clonal hematopoiesis. MF is recognized due to its distinct molecular profile (ie, presence of Janus kinase 2V617F [JAK2V617F], CALR, and myeloproliferative leukemia virus oncogene mutations), hematologic hypo- or hyperproliferation, and capacity to transform into acute myeloid leukemia (AML). Initial treatment in MF is tailored to prognostic risk, splenomegaly, mitigating burdensome symptoms, and maintaining blood counts. The symptom burden reduction, quality of life improvement, reduction in splenomegaly, stabilization of bone marrow fibrosis, and increased overall survival with ruxolitinib have solidified its role as a first-line agent in symptomatic or intermediate- to high-risk MF. However, the clinical challenges accompanying patients who are initially refractory, relapsed, or intolerant to ruxolitinib or those who progress to blast-phase disease are becoming increasingly relevant as MF patients live longer. In this article, we detail our treatment paradigm for the challenging clinical scenarios of ruxolitinib-relapsed/refractory MF and blast-phase myeloproliferative neoplasm (BP-MPN).
Background
Natural history of MPN
MF can arise as a secondary process from antecedent essential thrombocythemia or polycythemia (termed secondary MF), or it can arise de novo as primary MF. Over the disease course, individuals with MF may experience worsening of cytopenias, infections, transfusion dependence, progressive splenomegaly, or transformation to AML. Mortality for MF patients is most often due to progression to secondary AML (17%), progressive bone marrow failure/transfusion dependence (10%), thrombosis (7%), infections (6%), bleeding (3%), or other secondary malignancies (2%).1
Genetics
MF exhibits many common overlapping genetic mutations with AML (Figure 1).2-4 This is particularly true for progressive and BP-MPN disease, where mutational profiles have been found to have high concordance with those seen in de novo AML, post-MPN AML, and myelodysplastic syndrome (MDS)5-7 ; complex of karyotype or multiple genetic mutations are risk factors for poor prognosis and disease progression.8 Genomic alterations in BP-MPN are 3-fold more abundant compared with in chronic-phase MPN (P < .001).9

Current first-line treatment paradigm and challenges
Available for treatment outside of the clinical setting in the United States since 2011, ruxolitinib remains the only Food and Drug Administration (FDA)–approved therapy for MF. In the recently developed National Comprehensive Cancer Network (NCCN) guidelines, ruxolitinib is suggested for use in low-risk symptomatic patients as well as intermediate- and high-risk patients, with consideration of bone marrow transplantation and clinical trial if applicable.9 Although originally approved for use due to 2 large phase 3 clinical trials showing symptom and spleen response, additional benefits may include overall survival benefit10 and stabilization or reversal of bone marrow fibrosis.11 In an independent, blinded review by 3 hematopathologists for consensus-based World Health Organization bone marrow histology grading, MF patients treated with ruxolitinib showed a greater chance of reversal or stabilization of bone marrow fibrosis compared with patients treated with the best available therapy.11 However, this bone marrow effect was not observed in all patients and is still of uncertain clinical significance. The survival advantage for upfront ruxolitinib has been found independent of a clinical trial setting, with analysis of 2 commercial claims databases in the United States finding a median overall survival rate of 30 months in ruxolitinib-treated MF patients compared with 22 months for other treatments (N = 6982; hazard ratio, 0.7; 95% confidence interval, 0.6-0.8).12 Overall, MF patients are likely living longer in the era of first-line ruxolitinib treatment. Despite these significant advancements, ruxolitinib is not curative for MF, and ∼50% of individuals discontinue ruxolitinib after 3 years, most commonly due to loss or diminished response; also, ruxolitinib has not been found to significantly alter the rate of transformation to AML.13 Rather, 35% of patients on ruxolitinib will experience clonal evolution associated with decreased overall survival (6 vs 16 months).14
Inadequate ruxolitinib response
Inadequate response to ruxolitinib represents a spectrum of intolerance and refractory/relapsed disease features related to the spleen and/or symptoms leading to inability to continue ruxolitinib for its clinical indication.
Ruxolitinib refractory
Generally, being ruxolitinib refractory suggests that one has lost a spleen or symptom response to single-agent ruxolitinib. Features associated with the greatest risk of loss of response to ruxolitinib are (1) a mean titrated ruxolitinib dose of ≤10 mg twice daily, (2) delay in ruxolitinib treatment start ≥2 years from diagnosis, (3) transfusion dependence, (4) platelet counts <200 × 109/L, (5) grade 3 bone marrow fibrosis, (6) large splenomegaly (spleen palpable ≥10 cm below the left costal margin), and (7) International Prognostic Scoring System Intermediate-2 or high risk.12
Optimizing ruxolitinib dosing.
For patients who can tolerate continued ruxolitinib dosing but have loss of symptom or spleen response, optimizing their ruxolitinib dose can often lead to reinduction of ruxolitinib benefit. For those with dose-limiting anemia leading to suboptimal ruxolitinib dosing, we typically combine ruxolitinib with erythropoietin or danazol. In a multicenter, phase 2 trial of ruxolitinib and danazol, this combination resulted in stabilization of anemia in some patients (55.5% of patients on prior ruxolitinib had hemoglobin stabilization or improvement, with 88.9% having stable or increased platelets).15 The combination of erythropoietin analogs and ruxolitinib seemed more effective, with an overall response rate of 87.9% and a median response duration of 31 months.16 Generally, patients will undergo an improvement in erythropoietin levels after ruxolitinib initiation,17 and therefore, we will typically assess erythropoietin level after a patient is on a stable dose of ruxolitinib and use a threshold of <500 mU/mL (as per current NCCN guidelines18 ) to help guide our selection of either danazol or erythropoietin analog. Outside the use of ruxolitinib, the threshold of an erythropoietin level <125 mU/mL has been recommended19 due to its association with an anemia response.20
Symptom-directed complementary therapies.
We also consider symptom-directed therapies that can be used safely in combination with ruxolitinib for individuals who have symptom needs despite the best tolerable dose of ruxolitinib therapy. Although this topic is beyond the scope of this review, we generally consider specific treatment of erythromelalgia symptoms (ie, aspirin), pruritus (ie, H1 and H2 blockers, aspirin, UV light, cromolyn sodium, hydroxyzine, and topical steroids), and fatigue (ie, exercise, yoga, psychostimulants, selective and serotonin reuptake inhibitors).
Ruxolitinib relapsed
Although clearly defined response criteria have been established for ruxolitinib,21 inadequate ruxolitinib response remains a spectrum of inconsistently defined features that lack consensus in clinical trials (Table 1).22-25 In a clinical setting, generally, we use a patient-centric definition of inadequate ruxolitinib response consisting of ruxolitinib administration for at least 2 months (if achievable) with at least 1 of the following: (1) loss or lack of spleen or symptom response that negatively impacts quality of life, (2) sustained red blood cell transfusion requirement or dose-limiting anemia despite anemia-directed combinational therapies (as mentioned above), or (3) need to adjust dose of ruxolitinib to subtherapeutic dosing for thrombocytopenia, sustained or severe bleeding, or hematoma. Although ruxolitinib dosing typically correlates to ruxolitinib response,12 patients may experience symptomatic benefit at doses as low as 5 mg twice daily.
Definitions of ruxolitinib failure used in phase 2 and 3 clinical trials
| Failure feature* | Definition | Phase 2/3 trial(s) |
|---|---|---|
| Ruxolitinib duration | At least 14 d (less if intolerability or allergy) | JAKARTA II (NCT01523171) |
| At least 28 d | SIMPLIFY 2 (NCT02101268) | |
| ≥3 mo (if initial response was observed) or ≥28 d (if transfusion, hemorrhage, or cytopenias) | PAC203 (NCT03165734) | |
| Ruxolitinib dose | <20 mg twice daily | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Spleen response | Increase in spleen volume of ≥25% from nadir, splenic irradiation or splenectomy | COMFORT II (NCT00934544) |
| <10% spleen volume reduction by magnetic resonance imaging or <30% decrease from baseline in spleen length by physical examination or regrowth to these parameters after an initial response | PAC203 (NCT03165734) | |
| Red blood cell transfusions | Required red blood cell transfusions (at least 2 U/mo for 2 mo in PAC203) | PAC203 (NCT03165734) |
| SIMPLIFY 2 (NCT02101268) | ||
| Thrombocytopenia | CTCAE grade ≥3 | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Anemia | CTCAE grade ≥3 | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Hemorrhage or hematoma | CTCAE grade ≥3 (or bleeding) | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Progression | Peripheral blood blast percentage ≥20% sustained for ≥8 wk or bone marrow blast count ≥20% | COMFORT II (NCT00934544) |
| Failure feature* | Definition | Phase 2/3 trial(s) |
|---|---|---|
| Ruxolitinib duration | At least 14 d (less if intolerability or allergy) | JAKARTA II (NCT01523171) |
| At least 28 d | SIMPLIFY 2 (NCT02101268) | |
| ≥3 mo (if initial response was observed) or ≥28 d (if transfusion, hemorrhage, or cytopenias) | PAC203 (NCT03165734) | |
| Ruxolitinib dose | <20 mg twice daily | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Spleen response | Increase in spleen volume of ≥25% from nadir, splenic irradiation or splenectomy | COMFORT II (NCT00934544) |
| <10% spleen volume reduction by magnetic resonance imaging or <30% decrease from baseline in spleen length by physical examination or regrowth to these parameters after an initial response | PAC203 (NCT03165734) | |
| Red blood cell transfusions | Required red blood cell transfusions (at least 2 U/mo for 2 mo in PAC203) | PAC203 (NCT03165734) |
| SIMPLIFY 2 (NCT02101268) | ||
| Thrombocytopenia | CTCAE grade ≥3 | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Anemia | CTCAE grade ≥3 | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Hemorrhage or hematoma | CTCAE grade ≥3 (or bleeding) | SIMPLIFY 2 (NCT02101268) |
| PAC203 (NCT03165734) | ||
| Progression | Peripheral blood blast percentage ≥20% sustained for ≥8 wk or bone marrow blast count ≥20% | COMFORT II (NCT00934544) |
CTCAE, Common Terminology Criteria for Adverse Events.
Inadequate or recurrent symptom response criteria not defined in previous clinical trials, although symptom response has been defined as ≥50% reduction in the pretreatment Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score.21
For all patients with higher-risk disease features, we begin the discussion of allogeneic stem cell transplantation as soon as possible in the disease course (Figure 2), a conversation that typically occurs before a patient is ruxolitinib refractory or relapsed.26 Given worsened outcomes after ruxolitinib discontinuation, patients who are candidates would undergo transplant at the time of best response in their disease course and before ruxolitinib-relapsed or -refractory disease.27 Clinical trial options should be reviewed at the first signs of ruxolitinib-relapsed disease.
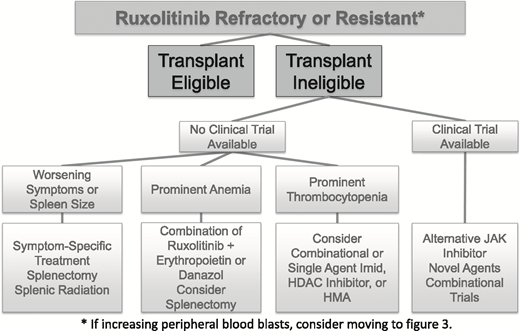
Commercially available off-label treatment options.
Alone or in combination with ruxolitinib, alternative commercially available treatments are available for MF with variable degrees of response. For individuals with early MF experiencing elevations in blood counts, interferon can be considered, although interferon can worsen symptom burden and risk of depression. Long-acting and more tolerable interferon agents are under investigation that have shown promising clinical activity in MF, including pegylated-interferon-α2a.28 Hypomethylating agents (HMAs) can be considered for alternative therapy, although their response rate seems to be highest in accelerated and blast-phase disease and carries risk of high-grade myelosuppression. In a general population of MPN patients, azacitidine (AZA) showed a response rate of 25% at 5 months, although 29% experienced grades 3 to 4 neutropenia.29 The combination of HMAs with ruxolitinib seems promising compared with single-agent ruxolitinib, with clinical trials of combinational ruxolitinib and AZA (starting after cycle 4) showing high response rates (72%) and spleen response (79%).30 Due to this excellent response rate, the combination of ruxolitinib and HMA is our preferred treatment out of the commercially available treatment options. The histone deacetylase (HDAC) inhibitor panobinostat, which is currently FDA approved for use in multiple myeloma, can also be considered for use in MF populations on an off-label basis. In a phase 2 trial of panobinostat, the response rate was 36%, with spleen reduction in 34%.31 Trials of the combination of panobinostat and ruxolitinib are ongoing (NCT01693601). Lenalidomide as a single agent has a 23% response rate with an improvement in anemia (19%) and/or spleen size (10%), although dose- and treatment-limiting effects can include myelosuppression (88% with at least grade 3 hematologic toxicity).32 We generally avoid the combination of ruxolitinib with lenalidomide due to excessive toxicities, because almost one-half of patients undergoing this combination in a clinical trial setting experienced dose interruptions and discontinuation within 3 months.33
Ruxolitinib holiday.
There is also evidence that suggests that a ruxolitinib holiday may be effective in reacquisition of drug sensitivity in some ruxolitinib-refractory patients.34 Heterodimerization with other JAKs occurring during persistent JAK activation has been shown to be reversible after a drug holiday in vitro models.34 In a case series of 13 MF patients who experienced an inadequate response or loss of response to ruxolitinib, the majority of patients experienced improvement in constitutional symptoms and spleen size with ruxolitinib rechallenge (92% and 69%, respectively). The mean duration of time off of ruxolitinib therapy was 8 weeks (range, 3-176 weeks).35 Although the optimal duration of holiday is unknown and this strategy has not yet been tested in a clinical trial setting, a ruxolitinib holiday could be considered for some patients who can tolerate a lapse in active treatment. However, such strategies should be considered cautiously due to the risk of symptom rebound after abrupt cessation of JAK inhibitor therapy.
Spleen-directed treatment options.
Splenectomy can be considered in instances of prominent anemia, where the suspected cause is massive splenomegaly or when a patient is severely symptomatic due to splenomegaly with no alternative treatment options (ie, likely have failed at least 2 JAK inhibitors, including ruxolitinib). Given the increased morbidity and mortality associated with this procedure, we typically are very selective for a population that has limited comorbidities and lacks alternative treatment options.36 Splenic radiation can be helpful in select cases, although it is generally avoided due to risk of prolonged myelosuppression.36
Investigational trials
In addition to JAK-targeted novel therapeutic agents, numerous ongoing clinical trials are investigating other molecular targets either as single agents or in combination with ruxolitinib (Table 2). We typically consider a clinical trial option first before using commercially available drug alternatives or combinations.
Novel agents being investigated in MF
| Agent name | Mechanism of action | NCT identifier; references |
|---|---|---|
| Novel single-agent investigations*(or both as single agent and combined with ruxolitinib) in MF | ||
| ABT-888* | PARP inhibitor | 61 |
| Alisertib | Aurora kinase inhibitor | NCT02530619; 62 |
| Durvalumab | Anti-PDL1 antibody | NCT02871323 |
| Glasdegib* | Hedgehog pathway inhibitor | NCT02226172; 63 |
| Imetelstat | Telomerase inhibitor | NCT01731951; NCT0242608; 64 |
| LCL 161 | SMAC inhibitor | NCT02098161; 65 |
| OGX-427 | Heat shock protein | 66 |
| Mirabegron | Sympathicomimetic agonist | NCT02311569; 67 |
| Nivolumab | Anti-PD1 antibody | NCT02421354 |
| Pembrolizumab* | Anti-PD1 antibody | NCT03065400 |
| PRM151* | Antifibrosing agent | NCT01981850; 68 |
| Pracinostat | HDAC inhibitor | NCT01200498; NCT02267278; 69 |
| SL-401 | Recombinant IL-3 with diphtheria toxin | NCT02268253; 70 |
| Sotatercept* | ACTRIIa | NCT01712308; 71 |
| Umbralisib* | PI3K inhibitor | NCT01730248; 72 |
| Novel combinational investigations with ruxolitinib in MF | ||
| Vismodegib | Hedgehog pathway inhibitor | NCT02593760; 73 |
| PIM447, LEE001, and JAK1/2 | Pan-PIM inhibitor, CDK4/6 inhibitor, JAK1/2 inhibitor | NCT02370706; 74 |
| Sonidegib | Hedgehog pathway inhibitor | NCT01787552; 75 |
| JAK2 inhibitor investigations (published phase 3 investigations and above only) | ||
| Fedratinib | JAK2 inhibitor | NCT00724334; 37,38 |
| Momelotinib | JAK1/2 inhibitor | NCT01969838; NCT02124746; NCT03441113; 42 |
| Pacritinib | JAK2, FLT3 inhibitor | NCT03165734; 41,76 |
| Agent name | Mechanism of action | NCT identifier; references |
|---|---|---|
| Novel single-agent investigations*(or both as single agent and combined with ruxolitinib) in MF | ||
| ABT-888* | PARP inhibitor | 61 |
| Alisertib | Aurora kinase inhibitor | NCT02530619; 62 |
| Durvalumab | Anti-PDL1 antibody | NCT02871323 |
| Glasdegib* | Hedgehog pathway inhibitor | NCT02226172; 63 |
| Imetelstat | Telomerase inhibitor | NCT01731951; NCT0242608; 64 |
| LCL 161 | SMAC inhibitor | NCT02098161; 65 |
| OGX-427 | Heat shock protein | 66 |
| Mirabegron | Sympathicomimetic agonist | NCT02311569; 67 |
| Nivolumab | Anti-PD1 antibody | NCT02421354 |
| Pembrolizumab* | Anti-PD1 antibody | NCT03065400 |
| PRM151* | Antifibrosing agent | NCT01981850; 68 |
| Pracinostat | HDAC inhibitor | NCT01200498; NCT02267278; 69 |
| SL-401 | Recombinant IL-3 with diphtheria toxin | NCT02268253; 70 |
| Sotatercept* | ACTRIIa | NCT01712308; 71 |
| Umbralisib* | PI3K inhibitor | NCT01730248; 72 |
| Novel combinational investigations with ruxolitinib in MF | ||
| Vismodegib | Hedgehog pathway inhibitor | NCT02593760; 73 |
| PIM447, LEE001, and JAK1/2 | Pan-PIM inhibitor, CDK4/6 inhibitor, JAK1/2 inhibitor | NCT02370706; 74 |
| Sonidegib | Hedgehog pathway inhibitor | NCT01787552; 75 |
| JAK2 inhibitor investigations (published phase 3 investigations and above only) | ||
| Fedratinib | JAK2 inhibitor | NCT00724334; 37,38 |
| Momelotinib | JAK1/2 inhibitor | NCT01969838; NCT02124746; NCT03441113; 42 |
| Pacritinib | JAK2, FLT3 inhibitor | NCT03165734; 41,76 |
ACTRIIa, activin A receptor type IIA; CDK, cyclin-dependent kinase; FLT3, FMS-like tyrosine kinase 3; IL-3, interleukin-3; PARP, polyadenosine 5′-diphosphate-ribose polymerase; PD1, programmed T-cell death 1; PDL1, programmed death-ligand 1; PI3K, phosphatidyl 3-kinase; PIM, proto-oncogene serine/threonine-protein kinase.
Designates that this is being investigated both alone and in combination with ruxolitinib.
Alternative JAK inhibitors.
Since the approval of ruxolitinib in 2011, no new JAK inhibitors have made it through the FDA approval process for MPN, although there remains promising ongoing investigation. Fedratinib, a selective JAK2 inhibitor, elicited responses in 47% (400 mg) and 50% (500 mg) of patients with Intermediate-2 or high-risk MF and showed superiority to placebo for control of splenomegaly and symptoms in the JAKARTA I Trial.37 In the JAKARTA II Trial, fedratinib showed activity in the second-line setting for individuals who had previously been on ruxolitinib.38 In November 2013, fedratinib was placed on an FDA clinical hold due to concerns of Wernicke’s encephalopathy. However, in a recent analysis of the 8 of 877 treated patients with suspected cases of Wernicke’s encephalopathy, only 1 case was found to have Wernicke’s encephalopathy, and it predated the patient’s fedratinib use.39 Given this, the FDA lifted the clinical hold for fedratinib, and it is now undergoing clinical development.40 Pacritinib was evaluated in the PERSIST 141 and PERSIST 224 studies and is currently undergoing additional evaluation in a randomized, phase 3 dose optimization study (PAC203; NCT03165734). In the fall of 2017, the SIMPLIFY 142 and SIMPLIFY 225 trials of momelotinib were placed on hold due to inability to meet primary end points. Clinical trials of these alternative JAK2 inhibitors have shown reinduction of eliciting symptom benefit, and these agents remain a good treatment option in the ruxolitinib-refractory setting.
Novel targeted agents.
With the success of therapeutic targeting of the JAK-STAT axis, focus has turned to drugging downstream signaling pathways that are dysregulated in MF. These treatments, used either as single agents or in combination with ruxolitinib, target ligands of pathway activation, including the hedgehog, phosphatidyl 3-kinase (PI3K), proto-oncogene serine/threonine-protein kinase, cyclin-dependent kinase 4/6, polyadenosine 5′-diphosphate-ribose polymerase (PARP), aurora kinase, SMAC, and heat shock protein pathways. Other specific targets under investigation include antifibrosing agents (PRM151), epigenetic modification (HDAC inhibitors), telomerase inhibition (imetelstat), and sympathicomimetic signaling (mirabegron).
Immune modulation.
Ruxolitinib intolerant
Some individuals will be unable to tolerate ruxolitinib for alternative reasons (ie, other side effects, severe allergic reactions, and personal preference). We typically consider patients who are unable to tolerate a dose of ruxolitinib above 5 mg twice daily due to nonhematologic complications intolerant to ruxolitinib as well. In these individuals, our treatment algorithm is the same as in patients who relapsed on ruxolitinib as described above.
Case 1, continued
Six months later, the patient presents to see you back in clinic. By adding erythropoietin to ruxolitinib, you were able to gain a response in hemoglobin and use a higher dose of ruxolitinib. He subsequently experienced improvements in spleen size and symptom response. However, blood work from this morning reveals an increased white blood cell of 17 × 109/L, hemoglobin of 9.0 g/dL, and platelets of 115 × 109/L. His differential now shows the presence of 13% peripheral blasts. He otherwise feels well and continues to be active. You perform a bone marrow biopsy in the office that reveals 11% blasts. He continues to feel well and stay active at his job, although notes a significant increase in fatigue. He wishes to know the best next treatment strategy given his progression to BP-MPN.
BP-MPN
With outcomes worse than standard AML, salvage therapies in accelerated and BP-MPN rely on the tolerability and availability of cytoreductive therapies and allogeneic stem cell transplantation. In a recent review of BP-MPN, median overall survival was 3.6 months, a statistic that has not altered over the last 15 years.46 The formal International Working Group for MF Research and Treatment definition of blast phase is defined as persistent elevation in peripheral blood or bone marrow blasts of 20% or more.47 A second but closely related entity with shortened overall survival is accelerated-phase MPN, defined as individuals with an elevation of peripheral or bone marrow blasts between 10% and 20%.48
Clinical clues that the patient may be progressing to accelerated or blast phase include the rapid onset of severe cytopenias, transfusion dependence, and constitutional symptoms. Our initial workup in this setting includes a repeat bone marrow biopsy with karyotype to help understand the extent of blast cell involvement. The reevaluation of next generation genetic sequencing, if not done recently, is also critical in this setting to determine whether targetable mutations exist.
Initial blast-phase treatment options
Despite the worsened survival of blast-phase patients compared with AML patients as mentioned above, current treatment options for blast-phase MF mirror closely the current treatment paradigm in AML (Figure 3).
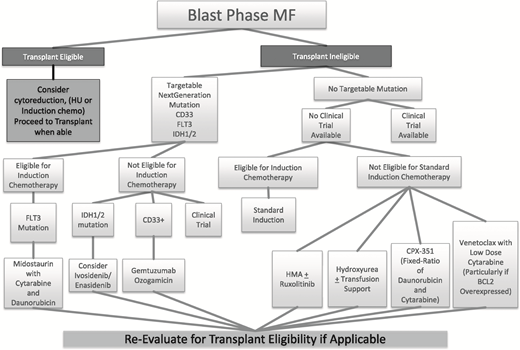
Treatment options for fit patients.
Although the antiquated “7 plus 3” induction with salvage allogeneic stem cell transplant is still the most widely used initial treatment option for fit individuals, the rapid growth of available therapies in AML has created the opportunity for more targeted and tolerable agents in BP-MPN outside of a clinical trial setting. Many of these therapies have been investigated in populations relevant to blast-phase MF, including therapy-related AML and AML with dysplastic changes.
Allogeneic stem cell transplantation in the blast-phase population remains the only curative treatment applicable to a select population of young and otherwise fit individuals. In general, fit patients are considered to have a biologic or physiologic age of <60 years, adequate left ventricular ejection fraction, near-normal pulmonary function testing, serum creatinine <1.5 mg/dL, serum bilirubin <2 mg/dL, liver function <2 times the upper limit of normal, and a body weight of 95% to 145% of ideal.49 However, even in this setting, outcomes are still suboptimal. In a recent analysis, survival rates at 5 years were 10% for patients who had achieved complete remission or incomplete count recovery and underwent subsequent allogeneic stem cell transplant.46 Despite this, transplant still remains the superior treatment option, with only 13% 5-year survival for similar patients who were not transplanted.46
Elderly, infirm, or relapsed BP-MPN patients.
For individuals who are not eligible for upfront traditional induction chemotherapy with allogeneic stem cell transplantation or clinical trials, we recommend an individualized approach incorporating disease burden, the best matching of treatment options via genetic sequencing, and prioritization of patient goals
HMAs (ie, AZA and decitabine), either alone or in combination with ruxolitinib as well as in combination with low-dose cytarabine, have also been used successfully in the setting of BP-MPN. As mentioned above, the combination of HMA with ruxolitinib appears to have very good response rates even in the setting of progressive disease. In 1 small case series of patients with BP-MPN, individuals experienced reduction in symptoms and splenomegaly and were able to attain disease stability with tolerable adverse events.50
There are 4 new approved agents that may have clinical utility in a BP-MPN population. The clinical agent CPX-351 (Vyxeos) is a new liposomal formulation that delivers a fixed ratio of daunorubicin and cytarabine, and it is approved for newly diagnosed treatment-related AML or AML with myelodysplasia-related changes. In patients 60 to 75 years of age with newly diagnosed treatment-related AML or AML with myelodysplasia-related changes, CPX-351 seemed to be more tolerable than standard cytarabine and daunorubicin induction.51 Given that BP-MPN seems genetically related to MDS, CPX-351 may represent a more tolerable agent and potentially, a therapeutically active agent in elderly BP-MPN patients.
Venetoclax (Venclexta) has received an FDA breakthrough therapy designation for use in combination with low-dose cytarabine in treatment-naïve elderly patients with AML who are ineligible for intensive chemotherapy. MF is a disease characterized by the overexpression of the antiapoptotic BCL-2 family of proteins (eg, BCL-XL and MCL-1).52 However, previous BCL-2 inhibitors (ie, Obatoclax)52 exhibited no significant single agent clinical activity in patients with MF at the dose and schedule evaluated. Despite this, venatoclax seems to be a promising combination treatment with potential efficacy in BP-MPN.
Enasidenib is a new agent approved for treatment of adults with relapsed or refractory AML with an isocitrate dehydrogenase-2 (IDH2) mutation.53 In this population, enasidenib was found to have an overall response rate of 40.3%, with median response duration of 5.8 months. Given that the frequency of IDH2 mutations is higher in the post-MPN AML (31%) and BP-MPN (21.6%; up to 44% if JAK2V617F is comutated) populations than in AML populations (12%)54 and that IDH mutations are associated with leukemic transformation (P < .01) and worsened survival (P = .01),55 this drug may have great clinical utility in the setting of blast-phase and post-MPN AML. The IDH1 inhibitor, ivosidenib, also may be a potential effective agent in post-MPN AML. Results from the phase 1 dose escalation and expansion single-arm trial of ivosidenib among a population of relapsed or refractory AML patients showed complete remission in 21.6% of treated patients with some complete molecular remissions.56 This study led to the FDA approval of ivosidenib for relapsed and refractory AML patients with the IDH1 mutation in July 2018. Until clinical trials show efficacy upfront in the use of IDH mutant populations, we suggest continuing to wait for the relapsed or refractory setting before initiating use in BP-MPN.
Gemtuzumab ozogamicin is approved for use in CD33-positive adult AML patients. Gemtuzumab showed efficacy in both the phase 2 single-arm study of AML patients in first relapse (ie, MyloFrance-157 ) and the randomized phase 3 study of AML patients who were not candidates for intensive chemotherapy (ie, AML-1958 ). Although MF patients are not typically CD33 positive, acute panmyelosis with MF, a specific subtype of MF, has been found to display this cellular marker.59 Additionally, other studies on MF patients have found that spleens of MF patients can contain populations of CD33 malignant hematopoietic stem cells.60 This suggests that, for those who are not candidates for intensive therapy or who do not have alternative treatment options and are CD33 positive, gemtuzumab should be considered.
For many patients, however, low-intensity clinical trials, supportive care, including transfusions for symptomatic support, or hospice may be appropriate. Emphasis should be given to the patient’s wishes regarding duration and intensity of care, with a frank discussion of end-of-life considerations, including caregiver burden. The value of palliative care should also be discussed with patients in efforts to assist with symptomatic support.
Conclusions
Over the 8 years since the initial approval of ruxolitinib, the first FDA-approved agent for the treatment of MF patients, our understanding and clinical expectations of JAK2 inhibition have deepened. The challenging clinical spectrum of ruxolitinib initial resistance, loss of response, and intolerance has become increasingly common. Alternative JAK inhibitors, novel compounds either alone or in combination with ruxolitinib (ie, inhibitors of the hedgehog, PARP, PI3K, and aurora kinase pathways), and immunotherapies are being investigated in the after single-agent ruxolitinib setting. MF progression to blast phase is associated with an aggressive course and very poor prognosis, with allogeneic stem cell transplant representing a risky but optimal treatment modality for those who are candidates. In the nontransplant setting, an individualized approach is optimal that takes into consideration disease features, targeted therapy where appropriate, and prioritization of patient goals.
Acknowledgments
The authors thank the many collaborators on the MPN Quality of Life Study Group for their continued efforts on behalf of progressing the care of patients with MPNs.
Correspondence
Ruben A. Mesa, Mays Cancer Center, University of Texas MD Anderson, University of Texas, 7979 Wurzbach Rd, San Antonio, TX 78249; e-mail: mesar@uthscsa.edu.
