
Abstract
June 2018 was the 20th anniversary of the clinical use of the first tyrosine kinase inhibitor (TKI), imatinib, for chronic myeloid leukemia. Since then, the change in prognosis for patients with this disease is one of the major success stories of modern cancer medicine. The dilemmas that face physicians and patients are no longer only those concerned with delaying inevitable progression to the terminal blastic phase or selecting the individuals most likely to benefit from allogeneic stem-cell transplantation; rather, they are now focused also on the choice of TKI, the management of comorbidities and adverse effects, strategies to improve quality of life, and the appropriateness of a trial of therapy discontinuation. Interestingly, with 4 TKIs approved for frontline use, the choice of initial therapy continues to cause controversy, a situation made more complicated by the tantalizing prospect of treatment-free remission. In this manuscript, we will explore the factors influencing this decision and try to provide a pragmatic and clinically applicable solution.
Learning Objectives
Understand the relative outcomes of treatment with the 4 TKIs currently available for first-line treatment in chronic myeloid leukemia
Appreciate the patient-related factors that can drive the choice of first-line therapy in chronic myeloid leukemia
Appreciate the drug-related factors that can drive the choice of first-line therapy in chronic myeloid leukemia
Introduction
The story of chronic myeloid leukemia (CML) is truly remarkable. Once a fatal disorder, it is now a chronic illness compatible with a normal life expectancy for the majority of patients.1,2 This remarkable advance has been achieved via a deep understanding of the molecular pathogenesis, the sequential development and use of increasingly potent targeted tyrosine kinase inhibitors (TKIs), the rigorous use of molecular monitoring to assess response (or lack/loss thereof), and the reinforcement of the need for compliance with daily therapy. The fact is that an individual newly diagnosed with CML in chronic phase in 2018 is more likely to die as a result of another medical condition than of his or her leukemia.3,4 CML continues to surprise, with numerous studies consistently reporting that ∼40% to 50% of individuals with deep and durable molecular responses (MRs) can safely discontinue therapy.5-9
Assessing success in CML
Irrespective of the drug chosen, continued prescription depends on a combination of efficacy and tolerability. A number of guidelines are now available10,11 that define groups of patients destined to do well or less well by the depth of remission obtained at particular time points after initiation of treatment (Table 1). It may be important to remember that these milestones are derived from the outcome of patients treated first line with imatinib and may not be exactly applicable to those who start on the more potent second generation of TKIs (2GTKIs). Although we know that the achievement of a 1-log reduction in tumor load (quantitative reverse transcription polymerase chain reaction [qRT-PCR] < 10% International Scale [IS]; MR1) at 3 months and of 2 logs (complete cytogenetic remission [CCyR]; qRT-PCR < 1% IS; MR2) at 6 months with imatinib therapy distinguishes a cohort of patients with an excellent prognosis from those with a poorer outcome,12,13 and that these somewhat arbitrary milestones can be applied to the use of 2GTKIs,14-16 it is entirely possible that these levels could and should be lower if the initial therapy is a 2GTKI. Recently, evidence from the German CML IV study identified that the achievement of a 3-log reduction in tumor load (MMR) confers an advantage in progression-free survival over the achievement of CCyR only, although the time by which MMR should be achieved is less clear.17 The ultimate confirmation of efficacy of treatment is the ability to discontinue the TKI indefinitely without clinical, cytogenetic, or molecular evidence of recurrence of CML, now referred to as treatment-free remission (TFR). The depth and duration of remission required to consider a potential study of treatment discontinuation are not yet defined, but the minimum practical recommendation from the Euro-SKI study is a 4-log reduction in disease burden (MR4) and duration of imatinib therapy of at least 5.8 years.8
European (ELN) and US (NCCN) guidelines for assessing responses and directing treatment changes
| Time, months | ELN | NCCN | ||||
|---|---|---|---|---|---|---|
| Optimal | Warning | Failure | Green* | Yellow† | Red‡ | |
| 3 | BCR-ABL1 ≤10% and/or Ph+ ≤35% | BCR-ABL1 >10% and/or Ph+ 36%-95% | Non-CHR and/or Ph+ >95% | BCR-ABL1 ≤10% | BCR-ABL1 >10% | Not defined |
| 6 | BCR-ABL1 <1% and/or Ph+ 0% | BCR-ABL1 1%-10% and/or Ph+ 1%-35% | BCR-ABL1 >10% and/or Ph+ >35% | BCR-ABL1 ≤10% | Not defined | BCR-ABL1 >10% |
| 12 | BCR-ABL1 ≤0.1% | BCR-ABL1 >0.1%-1% | BCR-ABL1 >1% and/or Ph+ >0% | BCR-ABL1 ≤1% | BCR-ABL1 >1%-10% | BCR-ABL1 >10% |
| >12 | BCR-ABL1 ≤0.1% | CCA/Ph− (−7 or 7q−) | Loss of CHR, loss of CCyR, confirmed loss of MMR§ mutations, CCA/Ph+ | BCR-ABL1 ≤0.1% | BCR-ABL1 >0.1%-1% | BCR-ABL1 >1% |
| Time, months | ELN | NCCN | ||||
|---|---|---|---|---|---|---|
| Optimal | Warning | Failure | Green* | Yellow† | Red‡ | |
| 3 | BCR-ABL1 ≤10% and/or Ph+ ≤35% | BCR-ABL1 >10% and/or Ph+ 36%-95% | Non-CHR and/or Ph+ >95% | BCR-ABL1 ≤10% | BCR-ABL1 >10% | Not defined |
| 6 | BCR-ABL1 <1% and/or Ph+ 0% | BCR-ABL1 1%-10% and/or Ph+ 1%-35% | BCR-ABL1 >10% and/or Ph+ >35% | BCR-ABL1 ≤10% | Not defined | BCR-ABL1 >10% |
| 12 | BCR-ABL1 ≤0.1% | BCR-ABL1 >0.1%-1% | BCR-ABL1 >1% and/or Ph+ >0% | BCR-ABL1 ≤1% | BCR-ABL1 >1%-10% | BCR-ABL1 >10% |
| >12 | BCR-ABL1 ≤0.1% | CCA/Ph− (−7 or 7q−) | Loss of CHR, loss of CCyR, confirmed loss of MMR§ mutations, CCA/Ph+ | BCR-ABL1 ≤0.1% | BCR-ABL1 >0.1%-1% | BCR-ABL1 >1% |
Adapted from European LeukemiaNet (ELN) guidelines (version 2013)10 and National Comprehensive Cancer Network (NCCN) guidelines (version 2.2018)11 with permission.
CCA, clonal chromosome abnormality; CCyR, complete cytogenetic remission; CHR, complete hematological response; MMR, major molecular remission; Ph, Philadelphia chromosome.
Equivalent to good response, where no treatment change is recommended.
Equivalent to suboptimal response, where switch to alternative TKI, Imatinib dose escalation or hematopoietic stem cell transplant may be considered, according to the clinical scenario.
Equivalent to treatment failure, which imposes switching to alternative TKI and evaluation of hematopoietic stem-cell transplantation.
In 2 consecutive tests, 1 of which with a BCR-ABL1 transcript level ≥1%.
Outcome of first-line therapies
Considerable data now confirm the long-term outcome of first-line imatinib, derived not only from the original phase 3 randomized study of imatinib vs interferon and cytosine arabinoside but also from all control arms of the phase 3 randomized studies of 2GTKI vs imatinib in newly diagnosed patients and the German CML IV study, which randomized patients to varying doses of imatinib with or without concomitant interferon (Table 2).15,16,18-25 Patients treated upfront with imatinib have probabilities of 65% to 70% for CCyR at 12 months, 50% to 55% for MMR at 2 years, and 45% to 50% for MR4 at 5 years. The data from these studies in terms of depth of response are invariably reported as cumulative incidences at annual intervals and provide little information regarding the durability of those responses, which would be the prerequisite for a trial of discontinuation. However, information derived from sequential laboratory results suggests that the proportion of patients achieving deep MRs with frontline imatinib continues to increase beyond 8 years.23,26
Response and survival outcomes of first- and second-line TKI therapy in the largest clinical trials
| Clinical trial | MR1 at 3 mo, % | CCyR/MR2 at 12 mo, % | MR3 at 12 mo, % | MR4 at 2 y, % | MR4.5 at 5 y, % | OS, % | |
|---|---|---|---|---|---|---|---|
| 5 y | 10 y | ||||||
| Imatinib 400 mg once daily | |||||||
| IRIS24 | — | 70.90 | 50.2 | — | 40.2 | 89 | 83.3 |
| CML IV23 | 68.5* | 67.5 | 36.7 | 31 | 49.4 | 94† | 80 |
| ENESTnd16 | 66.7 | 65 | 27 | 18 | 31 | 91.7 | — |
| DASISION15 | 64 | 72 | 28 | — | 33 | 90 | — |
| BELA20 | 65.4 | 68 | 27 | 3 (1 y) | — | — | — |
| BFORE25 | 57 | 66 | 36.9 | — | — | — | — |
| S0325-219 | — | 69 | 44 | — | — | 97 (3 y) | |
| SPIRIT18 | — | 58 | 38 | 21 | — | — | — |
| SPIRIT221 | — | 40.1 | 42.6 | — | — | — | — |
| Nilotinib 300 mg twice daily | |||||||
| ENESTnd16 | 90.6 | 80 | 55 | 39 | 54 | 93.7 | — |
| ENEST1st22 | 97 | 95 | 56.3 | 40.4 | — | 98.9 (2 y) | — |
| Dasatinib 100 mg once daily | |||||||
| DASISION15 | 84 | 83 | 46 | — | 42 | 91 | — |
| S0325-219 | — | 84 | 59 | — | — | 97 (3 y) | — |
| SPIRIT221 | — | 51 | 58.1 | — | — | — | — |
| Bosutinib 500 mg once daily; 400 mg once daily | |||||||
| BELA20 | 86 | 70 | 41 | 12 (1 y) | — | — | — |
| BFORE25 | 75.20 | 77.2 | 47.2 | — | — | — | — |
| Clinical trial | MR1 at 3 mo, % | CCyR/MR2 at 12 mo, % | MR3 at 12 mo, % | MR4 at 2 y, % | MR4.5 at 5 y, % | OS, % | |
|---|---|---|---|---|---|---|---|
| 5 y | 10 y | ||||||
| Imatinib 400 mg once daily | |||||||
| IRIS24 | — | 70.90 | 50.2 | — | 40.2 | 89 | 83.3 |
| CML IV23 | 68.5* | 67.5 | 36.7 | 31 | 49.4 | 94† | 80 |
| ENESTnd16 | 66.7 | 65 | 27 | 18 | 31 | 91.7 | — |
| DASISION15 | 64 | 72 | 28 | — | 33 | 90 | — |
| BELA20 | 65.4 | 68 | 27 | 3 (1 y) | — | — | — |
| BFORE25 | 57 | 66 | 36.9 | — | — | — | — |
| S0325-219 | — | 69 | 44 | — | — | 97 (3 y) | |
| SPIRIT18 | — | 58 | 38 | 21 | — | — | — |
| SPIRIT221 | — | 40.1 | 42.6 | — | — | — | — |
| Nilotinib 300 mg twice daily | |||||||
| ENESTnd16 | 90.6 | 80 | 55 | 39 | 54 | 93.7 | — |
| ENEST1st22 | 97 | 95 | 56.3 | 40.4 | — | 98.9 (2 y) | — |
| Dasatinib 100 mg once daily | |||||||
| DASISION15 | 84 | 83 | 46 | — | 42 | 91 | — |
| S0325-219 | — | 84 | 59 | — | — | 97 (3 y) | — |
| SPIRIT221 | — | 51 | 58.1 | — | — | — | — |
| Bosutinib 500 mg once daily; 400 mg once daily | |||||||
| BELA20 | 86 | 70 | 41 | 12 (1 y) | — | — | — |
| BFORE25 | 75.20 | 77.2 | 47.2 | — | — | — | — |
Refers to overall rate across the 4 arms.
Refers to imatinib-only arms.
In all of the randomized studies of imatinib vs a 2GTKI, it is clear that the 2GTKI induces deeper responses more rapidly than imatinib and probably in a higher proportion of patients.15,16 If the goal of therapy is to increase the number of patients eligible for a trial of treatment discontinuation, the treatment of choice at diagnosis should be a 2GTKI.
However, none of the randomized studies of imatinib vs a 2GTKI have shown a survival advantage for either drug at a minimum of 5 years of follow-up, although the results from ENESTnd suggest that patients treated with nilotinib are less likely to die as a result of CML than those receiving imatinib.16 If, however, the death rate is identical in the 2 arms, it is possible that some of the non–CML-related deaths are related to the use of the more potent 2GTKI and the higher incidence of serious adverse events. If true, the dilemma is that the use of frontline 2GTKIs may increase the number of patients able to stop therapy, but potentially at the expense of inducing or exacerbating concomitant illness in others.
Two additional issues require consideration. The first is that the choice of first-line therapy in patients destined for TFR may be irrelevant, and the only real difference observed would be the speed at which they reach the time point for consideration of a trial of discontinuation. Because a patient cannot act as his or her own control, one way of trying to solve this dilemma would be if the use of a 2GTKI could induce a sustained deep response and TFR in a patient who had not reached this level of response with imatinib. This has been addressed in trials of stopping 2GTKIs,7,9 but these results are difficult to interpret, given that most patients included in these studies received the 2GTKI for imatinib intolerance rather than resistance. The data that do exist suggest that successful treatment discontinuation is rarely possible in those with true imatinib resistance.
In contrast, studies of commencing nilotinib in patients in MMR but not MR4.5 with imatinib have shown not only that deeper responses can be established with nilotinib but also that ∼40% to 50% of patients can stop therapy at a later date.27 Of the 163 patients who commenced nilotinib in this study, 126 were eligible to enter the TFR stage. The median duration of imatinib treatment in this group was 23.5 months (range, 0.2-129 months), and it is possible that some of these patients would have achieved deep responses had they continued to receive imatinib. In contrast, the median duration of imatinib treatment in the 37 patients ineligible for a trial of TFR was 45.4 months (range, 1-119 months).
The second consideration is whether the use of upfront imatinib followed by rigorous and accurate molecular monitoring and an early change to a 2GTKI if milestones are not met (particularly qRT-PCR < 10% IS at 3 and < 1% at 6 months) would restore the same prognosis to patients that they would have had if they had received a 2GTKI as first-line therapy. The phase 2 studies of a 2GTKI for imatinib resistance demonstrate that CCyR can be achieved in ∼40% to 50% of patients, but these studies were performed some years ago, when the median duration of imatinib treatment before the change of therapy was 30 to 40 months (range, <1-194 months).28-30 Although intuition suggests that an earlier change to a more effective drug should be the optimal strategy for management, the possibility exists that the results of the original studies overestimated CCyR rates because patients with particularly poor prognoses experienced progression and may have died before the 2GTKI became available for use. The Tidel-2 study attempted to address this issue by changing patients from imatinib to nilotinib if early response milestones were not met, and it achieved outcomes similar to those seen in randomized studies of first-line 2GTKIs (rate of MR3 at 1 year, 62%), but the participants were not directly compared with a group of patients starting a 2GTKI in the first-line setting, so any interpretation of the results must be cautious.31 Furthermore, the starting dose of imatinib in this study was 600 mg, and this dose has been shown to induce MR3 more rapidly than the standard dose of 400 mg.23
Considerations for choice of frontline treatment
Patient-related factors
It seems obvious that the choice of first-line therapy should be driven by the goal of treatment, simplistically defined as either long-term disease control with good quality of life or TFR. In clinical practice, these choices are often not straightforward.
Although the best biomarker for survival is currently the response to therapy at 3, 6, and 12 months, which is obviously not available at diagnosis, there are some factors that can be used to inform this choice. Perhaps the most controversial is age, where a straightforward approach might be to aim for TFR in the young and for safe, effective disease control in the elderly. Unfortunately, even this is complicated by a relative paucity of data regarding the safety and efficacy of 2GTKIs in pediatric and young adult populations and the increasing prevalence of comorbidities occurring with age such that TFR might be a desirable aim to avoid additional adverse events in older patients.
There is evidence from pediatric data that children tend to present with seemingly more aggressive disease, with higher white-cell counts and larger spleens,32 and this leads to the question of whether the standard risk scores (Sokal, Hasford, European Treatment Outcome Study [EUTOS], and EUTOS Long-Term Survival), which consider younger age as a good prognostic feature, are applicable to children and young adults.33,34 CML is unusual in individuals age <25 years, so attempts to look specifically at their prognosis within larger studies has proved difficult, either because those age <18 years were excluded from the trials or because the small numbers render the subgroup analysis invalid. Within the German CML IV study, younger age was not considered an adverse prognostic factor.35
In adults, the use of prognostic scores is more appropriate. The Sokal score has been used most commonly to date, and although designed to predict outcome in patients treated with busulphan, it has proven useful in the TKI era.16,24,25,36 The problem remains that, on an individual basis, the score does not guarantee that a patient with a low score at diagnosis will not develop progression or that a patient with a high score will not respond well to imatinib. The 2 large commercial phase 3 randomized studies of imatinib vs dasatinib15 and imatinib vs nilotinib16,37 assessed outcome using the Hasford and Sokal scores, respectively. The Sokal score predicted achievement of CCyR, MMR, and progression-free survival (Table 3). Patients classified as low risk had similar CCyR, MMR, and OS rates with either imatinib or nilotinib, whereas those with high-risk disease had improved outcomes with nilotinib, suggesting that the latter group should be offered 2GTKIs as first-line therapy. The situation regarding intermediate Sokal scores is more complicated, with similar CCyR but lower MMR and OS rates in patients treated with imatinib rather than nilotinib. Here the benefit of an early 2GTKI should be balanced against the possibility of adverse events in those with preexisting comorbidities. Data related to the impact of the Hasford score on OS in the Dasision study have not been reported, although higher rates of MMR were seen in the low-risk group compared with the intermediate- and high-risk groups.15
Molecular and survival outcomes according to Sokal score at diagnosis: ENESTnd randomized trial results
| Score | Imatinib 400 mg once daily, % | Nilotinib 300 mg twice daily, % | ||||
|---|---|---|---|---|---|---|
| CCyR* | MR3† | 5-y OS | CCyR* | MR3† | 5-y OS | |
| Low | 90 | 62.5 | 100 | 91 | 76.7 | 97 |
| Intermediate | 85 | 54.5 | 88.5 | 87 | 75.2 | 93.8 |
| High | 72 | 38.5 | 84.2 | 81 | 66.7 | 88.8 |
| Score | Imatinib 400 mg once daily, % | Nilotinib 300 mg twice daily, % | ||||
|---|---|---|---|---|---|---|
| CCyR* | MR3† | 5-y OS | CCyR* | MR3† | 5-y OS | |
| Low | 90 | 62.5 | 100 | 91 | 76.7 | 97 |
| Intermediate | 85 | 54.5 | 88.5 | 87 | 75.2 | 93.8 |
| High | 72 | 38.5 | 84.2 | 81 | 66.7 | 88.8 |
Adapted with permission.16
OS, overall survival.
Cumulative response at 2 years.
Cumulative response at 3 years.
Most recently, the German CML IV study also looked at the role of Sokal in predicting outcome in patients who were randomized to varying doses of imatinib or imatinib plus additional therapy with interferon or cytosine arabinoside. Although the score was predictive, the investigators were able to derive an additional algorithm, the ELTS score, which seems to more accurately distinguish, in the context of TKI therapy, those with high-risk disease compared with the Sokal score.23 The proportion of patients classified as high risk is lower than that with the Sokal score, but these patients have a particularly poor prognosis. If the score can be validated in other studies, it is likely that it will be more widely adapted and will direct first-line therapy. The prognostic value of additional chromosomal abnormalities (ACAs) at diagnosis has long been controversial, although the acquisition of ACAs during the disease course remains a criterion for accelerated-phase disease. The role of ACAs at diagnosis was clarified recently with a classification that defines major- and minor-route abnormalities,38 where major-route changes correlate with inferior survival. Choosing a 2GTKI for these latter patients would be entirely justified. The impact of the type of BCR-ABL1 transcript has also been difficult to interpret. There are 2 common transcripts, e13a2 and e14a2; e13a2 occurs alone in 40% of patients, and e14a2 occurs alone or together with e13a2 in 59% (Michele Baccarani, Fausto Castagnetti, Gabriele Gugliotta, Gianantonio Rosti, Simona Soverini, and Markus Pfirrmann for the International BCR-ABL Study Group, manuscript submitted, September 2018). It is not entirely clear whether this is a true reflection of the disease biology or whether the presence of 1 or 2 transcripts is a feature of RT-PCR artifact. Using standard primers, the e14a2 PCR product is longer the than its e13a2 counterpart and might simply be subject to less efficient PCR amplification. Several studies have reported that patients expressing the e14a2 transcript have a better outcome in terms of achievement of milestones,39-41 but an impact on survival has been shown less consistently.42,43 Approximately 1% to 2% of patients have a variety of alternative transcripts; because patients with these transcripts are usually excluded from clinical trials as a result of the absence of a standardized RT-PCR assay to monitor response, the impact of such transcripts on outcome is more difficult to assess.
Although many groups have looked for genetic biomarkers of response, the results to date have been somewhat disappointing. The presence of polymorphisms in the drug transporter proteins (eg, ABC1, OCT1, and SCL22) has shown some promise but has not been convincingly validated.44-47 Measuring changes in messenger RNA and/or protein expression in OCT1 and ABC1 after treatment initiation is technically challenging, and its benefit over measuring the reduction in BCR-ABL1 transcript numbers is unclear. Next-generation sequencing (NGS) technology now allows the detection of low-level kinase domain mutations present at diagnosis, but the clinical significance of such mutations is less predictable. Early attempts to identify somatic mutations in genes other than BCR-ABL1 to predict outcome (which has proved a promising approach in other hematological malignancies) has so far been unrewarding. Although mutations in a number of candidate genes have been identified in advanced-phase disease,48 chronic-phase disease has proven less genetically unstable than anticipated.49 Using targeted NGS panels consisting of a smaller number of genes in samples from newly diagnosed patients, several groups have been able to link somatic mutations, either single mutations in genes such as ASXL1 or mutations present in >1 gene, with poor prognosis.50 As these techniques become more widely available and are applied initially in extreme responders, we hope to see more information emerge, but given their relative rarity and the excellent prognosis of CML, validation will prove difficult without the collaboration of large trial groups.
Drug-related factors
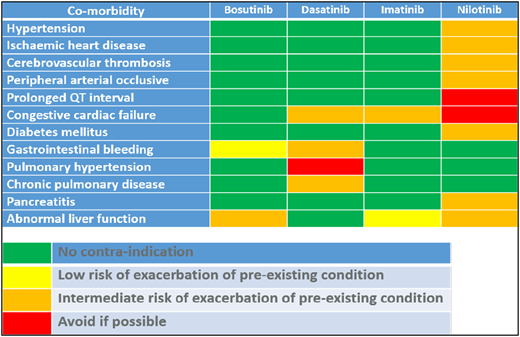
The currently available TKIs have a variety of adverse effects, some of which can be attributed to inhibition of off-target tyrosine kinases, and are common to all the drugs. However, each of the drugs has a particular adverse event profile that should be considered when choosing first-line therapy. The European LeukemiaNet recently reviewed the occurrence and management of TKI adverse effects,51 so the following discussion refers to adverse effects only in the context that they might direct the choice of drug.
Imatinib is generally regarded as the safest of the TKIs, having been in use for >20 years, with no long-term irreversible adverse effects described to date. In contrast, the 2G and 3GTKIs have individual risk profiles with more serious complications. The most severe and not necessarily fully reversible adverse effect of dasatinib is pulmonary arterial hypertension.52 Dasatinib is well known to cause pleural effusions in ∼25% to 30% of patients. Most effusions can be managed by temporary cessation of dasatinib until resolution, with reintroduction at the original dose followed by a reduction in dose if the effusion recurs. The first occurrence can be at any time postinitiation of therapy, and the incidence increases with age such that patients age >65 years have a chance of developing pleural effusion >60%.15 Other predisposing factors have been described, including prior autoimmune disease, prior occurrence of skin rash with imatinib or dasatinib, and history of hypertension and/or ischemic heart disease.53 Although prior respiratory disease has not been implicated in the risk factors for pulmonary arterial hypertension or pleural effusion, avoidance of further respiratory compromise is a sensible approach in a patient with known pulmonary disease. Both dasatinib and ponatinib affect platelet function,54 and hemorrhagic gastrointestinal colitis has also been described with dasatinib,55 so they may also be unsuitable for those with preexisting inflammatory bowel disease.
Nilotinib is associated with a low incidence of pancreatitis and would be contraindicated in patients with a history of pancreatitis and/or alcohol excess. More frequent and relatively common adverse effects include exacerbation of preexisting or new occurrence of diabetes mellitus, hypercholesterolemia, hypertension, or arterial occlusive events (AOEs) including myocardial infarction, cerebrovascular thrombosis, and peripheral arterial occlusive disease.16 The incidence of AOEs increases with increasing exposure to nilotinib and is predicted to reach >20% at 10 years. As a consequence, nilotinib is a less attractive choice for the newly diagnosed patient with diabetes, hypercholesterolemia, and/or preexisting arterial disease, and caution should be exercised in those with obesity, family history of ischemia, or history of smoking.
Bosutinib is associated with gastrointestinal toxicity, predominantly diarrhea. Fortunately, this is usually temporary. A more problematic adverse effect is drug-induced hepatitis, so attention should be paid to a history of liver disease and excess alcohol consumption.
Ponatinib is the most potent TKI to date, but its use as a first-line agent was curtailed when the phase 3 randomized study of ponatinib vs imatinib was halted prematurely because of a disturbingly high incidence of AOEs in patients receiving ponatinib compared with those receiving imatinib.56 This confirmed earlier findings in the phase 2 studies of either first-line ponatinb57 or when it was given for failure of prior TKIs.58 The manufacturer has not pursued a first-line indication in chronic-phase disease, but the upfront use of ponatinib together with intensive IV chemotherapy is currently being explored in blast crisis.
Finally, when advising on first-line treatment, an important factor is the lifestyle of the individual patient. Chronic drug use is associated with poor compliance, and helping the patient to fit treatment into his or her daily pattern will minimize this risk. Imatinib and bosutinib should be taken once daily after meals, and nilotinib twice daily before and after periods of fasting. Dasatinib can be taken before or after eating. Importantly, approximately a significant minority of women of child-bearing age, 25% in 1 study,59 wish to have children after their diagnosis. All these drugs are contraindicated in pregnancy, and ideally, the woman should stop therapy before conception. This is best done when the individual has achieved the same minimum milestones that would render her eligible for a trial of TFR, namely MR4 for at least 12 months. Because this goal is most rapidly achieved using a 2GTKI, this could be an argument for recommending the more potent therapies as first-line treatment in this group.
How can we make the choice in clinical practice?
Firstly, it is hard to go wrong with imatinib. Not only does the drug have a 2-decade history of efficacy and safety, but it is now widely available in generic form with a concomitant reduction in cost. Adherence to current guidelines and the use of molecular monitoring should ensure that most patients destined to fare poorly with imatinib will be recognized within the first 3 to 6 months and offered more potent therapy. In all of the randomized studies of imatinib vs a 2GTKI, trial eligibility included a time from diagnosis to treatment of up to 3 months, and there has been no suggestion that longer times from diagnosis to start of therapy are associated with poorer outcome.
However, we are also aware that in the TKI era, patients tend to experience progression from chronic phase early in their disease course and often in the first year of treatment. If we are to try to make a difference in outcome for this small but important minority of patients, we require better biomarkers of their prognosis so that they can be offered more potent therapy and rapid referral for transplantation if early milestones are not reached. It is a sobering thought that even this approach may prove unsuccessful, but this is not a reason not to make every effort to improve their outcome. Although we were hopeful that the newer molecular technologies of NGS, whole-exome and whole-genome sequencing, transcriptomics, and epigenetics would identify markers of prognosis, we do not have any test that currently can be used in clinical practice. In the meantime, the best surrogates remain ACAs at diagnosis and high-risk classification in the Sokal and ELTS systems. These patients are probably best served by treatment with 2GTKIs from diagnosis, unless their comorbidities render the balance of risk between avoidance of early progression and harm through exacerbation of prior illnesses in favor of imatinib.
An argument commonly used to support initial therapy with imatinib for all patients is cost, because generic versions are seemingly equally effective and much less expensive than the original Novartis product and all the 2GTKIs. Indeed, in many countries, the cost of 2GTKIs precludes their approval for first-line use. Where 2GTKIs are readily available, it is worth remembering that the major cost over the lifetime of an individual will lie in the drug used for the longest period of time. Because approximately half of the patients treated initially with imatinib will have changed their drug at least once 8 years later and will presumably remain on their second or subsequent choice of drug for the rest of their lives, this argument becomes less convincing. Furthermore, if the use of a 2GTKI first line increases the proportion of patients able to discontinue therapy and allows them to stop treatment earlier than if they were treated with imatinib, the economic balance for the health care provider will be altered. Finally, 2GTKIs will all come off patent at some point in the future, underlining the need to make decisions regarding first-line therapy on biological rather than economic grounds.
The argument to use imatinib for all patients with low- or intermediate-risk disease is weaker if the goal of treatment is TFR. As discussed earlier, it is possible that the patient destined for successful, as opposed to a trial of, discontinuation will reach this goal irrespective of the initial treatment. However, there seems to be little doubt that TFR will be achieved more rapidly using a 2GTKI as initial therapy. The concern is that the widespread adoption of first-line 2GTKIs may increase the number of patients offered trials of TFR within the early years of diagnosis but that this benefit for some will be offset by harm and even premature death in others. For the patient with no comorbidities at diagnosis, and in countries where the drugs are not only available but cost considerations are less impactful, use of a 2GTKI upfront is entirely reasonable. If the use of these drugs is complicated by adverse effects, an as yet untried approach, at least in clinical trials, is to return the patient to imatinib once a deep MR is achieved. The obvious implication of such a therapeutic strategy is that younger patients will receive 2GTKIs and a majority of older patients will be treated with imatinib, but the use of specific age cutoffs is less useful than a holistic assessment of patient health.
In practice, the decision will be taken after an assessment of the patient and his or her disease and comorbidities, a discussion regarding the risks and benefits of each drug, and a frank conversation regarding his or her personal treatment goals. Guidance such as that summarized in Table 4 can assist both patient and physician, and whatever the initial choice, ongoing care mandates rigorous molecular monitoring, arranging for medical care of any comorbid disease, and managing adverse events promptly and effectively. Fortunately, for a great majority of patients, there will be an appropriate TKI.
Acknowledgments
J.F.A. is a National Institute for Health Research (NIHR) Senior Investigator and acknowledges the support of the Imperial College NIHR Biomedical Research Centre.
Correspondence
Jane F. Apperley, Centre for Haematology, Hammersmith Hospital, Du Cane Rd, London W12 0NN, United Kingdom; e-mail: j.apperley@imperial.ac.uk.
