
Abstract
Juvenile myelomonocytic leukemia (JMML) is a unique clonal hematopoietic disorder of early childhood characterized by hyperactivation of the RAS signal transduction pathway. Approximately 90% of patients harbor molecular alteration in 1 of 5 genes (PTPN11, NRAS, KRAS, NF1, CBL), which define genetically and clinically distinct JMML subtypes. Three subtypes, PTPN11- , NRAS-, and KRAS-mutated JMML, are characterized by heterozygous somatic gain-of-function mutations in non syndromic children, while two subtypes, JMML in neurofibromatosis type 1 and in JMML in children with CBL syndrome, are characterized by germ line RAS disease and acquired biallelic inactivation of the respective tumor suppressor genes in hematopoietic cells. In addition to the initiating RAS pathway lesion, secondary genetic alterations within and outside of the RAS pathway are detected in about half the patients. Most recently, genome-wide DNA methylation profiles identified distinct methylation signatures correlating with clinical and genetic features and highly predictive of outcome. JMML is a stem cell disorder, and most JMML patients require allogeneic stem cell transplantation for long-term survival. However, spontaneous disease regression is noted in the majority of children with CBL-mutated JMML and in some NRAS-mutated cases. In the absence of 1 of the 5 canonical RAS pathway alteration, rare mutations in other RAS genes and non-JMML myeloproliferative disorders need to be excluded. Understanding the genetic basis of myeloproliferative disorders in early childhood will greatly improve clinical decision making.
Learning Objectives
Understand that JMML is a RAS-driven myeloproliferative/myelodysplastic clonal neoplasia with 5 distinct genetic subtypes
Recognize JMML patients that may experience long-term survival in the absence of hematopoietic stem cell transplantation
Appreciate that DNA methylation status is a better predictor of clinical outcome than JMML genetic subgroup
Learn about other myeloproliferative disorders presenting in early childhood
Definition, clinical, and hematological description of JMML
Juvenile myelomonocytic leukemia (JMML) is a clonal hematopoietic disorder of childhood characterized by proliferation principally of the granulocytic and monocytic lineages.1 The age at diagnosis ranges from 1 month to early adolescence, but about one-half of the cases occur in children <2 years of age.2 Splenomegaly is present in virtually all cases, and hepatomegaly, lymphadenopathy, and skin rashes are common. Dry cough, tachypnea, and interstitial infiltrates on chest radiograph are signs of peribronchial and interstitial pulmonary infiltrates, whereas gut infiltrates may predispose to bloody diarrhea and gastrointestinal infections. JMML very rarely involves the central nervous system.
On peripheral blood (PB) smear, leukocytosis, monocytosis, and presence of immature monocytes, along with myelocytes, metamyelocytes, and nucleated red cells, are usually evident. Often, a few blasts can be noted. Thrombocytopenia and anemia are commonly present. Bone marrow (BM) findings in JMML are by themselves not diagnostic, but rather are consistent with the diagnosis. The marrow blast count can be moderately elevated, but does not reach the level seen in acute leukemia. Megakaryocytes are reduced in number or absent in about two third of cases.
Chromosomal studies of leukemic cells show monosomy 7 in ∼25% of JMML patients, with other abnormalities in 10%, but the majority of patients (65%) have a normal karyotype.2 A remarkable feature of many JMML cases is increased synthesis of hemoglobin F (HbF). Signs of autoimmunity such as antinuclear antibodies or positive anti-globin test can be noted in up to 25% of children with JMML, whereas hypergammaglobulinemia is present in more than one-half of them.2
Genetic subtypes of JMML
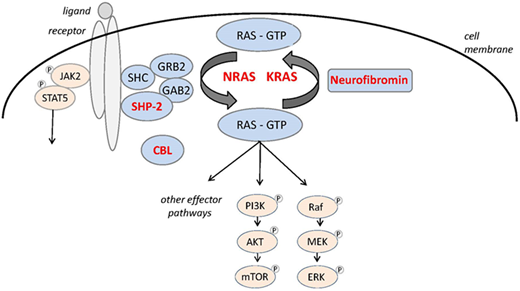
Altered RAS signaling is the main driving event in JMML. About 90% of patients harbor molecular alteration in 1 of 5 genes (PTPN11, NRAS, KRAS, CBL, and NF1) that encode proteins that when mutated activate RAS effector pathways (Figure 1; Table 1).1 The 5 canonical RAS pathway alterations describe 5 genetically and clinically distinct JMML subtypes. Three subtypes, PTPN11‐, NRAS‐, and KRAS‐mutated JMML, are characterized by heterozygous somatic gain‐of‐function mutations in nonsyndromic children, whereas 2 subtypes, JMML in neurofibromatosis type 1 (NF1, MIM 162200) and JMML in children with CBL syndrome (MIM 613563), are characterized by germ line RAS disease (RASopathy) and acquired biallelic inactivation of the respective NF1 or CBL tumor suppressor gene in hematopoietic cells. Because of a broad overlap in clinical and hematological presentation (Table 2), the classification of JMML into its 5 subtypes requires molecular workup.
Genetic JMML subtypes
| Genetic subtype | Patient cohort | ||
|---|---|---|---|
| Japanese (N = 135), %* | North American (N = 98), %† | EWOG‐MDS (N = 175), %‡ | |
| NF1 | 5 | 13 | 10 |
| PTPN11 | 36 | 32 | 41 |
| KRAS | 17 | 14 | 14 |
| NRAS | 15 | 17 | 16 |
| CBL | 17 | 11 | 12 |
| Negative | 9 | 12 | 7 |
Clinical and hematological characteristics of JMML genetic subtypes at diagnosis
| CBL (N = 43) | NF1 (N = 51) | PTPN11 (N = 180) | KRAS (N = 68) | NRAS (N = 65) | P | ||
|---|---|---|---|---|---|---|---|
| Clinical | Age (y), median (range) | 0.9 (0.1‐5.0) | 2.8 (0.3‐12.3) | 2.1 (0.1‐16.3) | 0.9 (0.1‐10.0) | 1.2 (0.1‐13.2) | <.01 |
| Male sex, % | 56 | 65 | 60 | 75 | 74 | .07 | |
| Spleen (palpable cm ↓ CM), median (range)* | 5 (0‐15) | 4 (0‐15) | 4 (0‐13) | 5 (0‐15) | 5 (0‐15) | NS | |
| PB | WBC (×109/L), median (range) | 27 (8‐113) | 32 (3‐131) | 30 (2‐424) | 21 (4‐177) | 41 (5‐312) | <.01 |
| Blasts (%), median (range) | 0 (0‐9) | 2 (0‐25) | 2 (0‐21) | 2 (0‐21) | 2 (0‐18) | <.01 | |
| Precursors (%), median (range)† | 4 (0‐42) | 5 (0‐31) | 9.5 (0‐56) | 7 (0‐65) | 8.2 (0‐45) | .05 | |
| Eosinophils (%), median (range) | 2 (0‐8) | 2 (0‐15) | 1 (0‐26) | 2 (0‐9) | 2 (0‐18) | .01 | |
| AMC (×109/L), median (range) | 4.0 (1‐48) | 7.6 (1‐26) | 4.1 (0‐237) | 5.6 (0‐40) | 6.2 (9‐232) | <.01 | |
| Platelet count (×106/L), median (range) | 92 (14‐289) | 129 (15‐357) | 50 (5‐373) | 42 (10‐182) | 60 (9‐232) | <.01 | |
| Hemoglobin (g/100 mL), median (range) | 9.0 (5‐13) | 10.0 (4‐13) | 9.1 (3‐13) | 8.7 (4‐14) | 8.9 (4‐13) | NS | |
| HbF (%), median (range) | 4 (0‐59) | 23 (1‐75) | 18 (0‐100) | 10 (0‐94) | 13 (0‐70) | <.01 | |
| BM | MP:EP, median (range) | 5.4 (1.1‐45) | 5.7 (0.4‐23) | 3.7 (0.1‐233) | 2.9 (0.3‐219) | 3.5 (0.5‐97) | <.01 |
| Blasts (%), median (range) | 3 (0‐20) | 5 (0‐27) | 4 (0‐34) | 4 (0‐20) | 4 (0‐20) | NS | |
| Eosinophils (%), median (range) | 2 (0‐15) | 3 (0‐12) | 2 (0‐33) | 3 (0‐18) | 2 (0‐18) | NS | |
| Karyotype | Normal (%) | 97 | 77 | 69 | 40 | 86 | <.01 |
| Monosomy 7 (%) | 0 | 13 | 21 | 52 | 7 | ||
| Other aberration (%) | 3 | 10 | 10 | 8 | 7 |
| CBL (N = 43) | NF1 (N = 51) | PTPN11 (N = 180) | KRAS (N = 68) | NRAS (N = 65) | P | ||
|---|---|---|---|---|---|---|---|
| Clinical | Age (y), median (range) | 0.9 (0.1‐5.0) | 2.8 (0.3‐12.3) | 2.1 (0.1‐16.3) | 0.9 (0.1‐10.0) | 1.2 (0.1‐13.2) | <.01 |
| Male sex, % | 56 | 65 | 60 | 75 | 74 | .07 | |
| Spleen (palpable cm ↓ CM), median (range)* | 5 (0‐15) | 4 (0‐15) | 4 (0‐13) | 5 (0‐15) | 5 (0‐15) | NS | |
| PB | WBC (×109/L), median (range) | 27 (8‐113) | 32 (3‐131) | 30 (2‐424) | 21 (4‐177) | 41 (5‐312) | <.01 |
| Blasts (%), median (range) | 0 (0‐9) | 2 (0‐25) | 2 (0‐21) | 2 (0‐21) | 2 (0‐18) | <.01 | |
| Precursors (%), median (range)† | 4 (0‐42) | 5 (0‐31) | 9.5 (0‐56) | 7 (0‐65) | 8.2 (0‐45) | .05 | |
| Eosinophils (%), median (range) | 2 (0‐8) | 2 (0‐15) | 1 (0‐26) | 2 (0‐9) | 2 (0‐18) | .01 | |
| AMC (×109/L), median (range) | 4.0 (1‐48) | 7.6 (1‐26) | 4.1 (0‐237) | 5.6 (0‐40) | 6.2 (9‐232) | <.01 | |
| Platelet count (×106/L), median (range) | 92 (14‐289) | 129 (15‐357) | 50 (5‐373) | 42 (10‐182) | 60 (9‐232) | <.01 | |
| Hemoglobin (g/100 mL), median (range) | 9.0 (5‐13) | 10.0 (4‐13) | 9.1 (3‐13) | 8.7 (4‐14) | 8.9 (4‐13) | NS | |
| HbF (%), median (range) | 4 (0‐59) | 23 (1‐75) | 18 (0‐100) | 10 (0‐94) | 13 (0‐70) | <.01 | |
| BM | MP:EP, median (range) | 5.4 (1.1‐45) | 5.7 (0.4‐23) | 3.7 (0.1‐233) | 2.9 (0.3‐219) | 3.5 (0.5‐97) | <.01 |
| Blasts (%), median (range) | 3 (0‐20) | 5 (0‐27) | 4 (0‐34) | 4 (0‐20) | 4 (0‐20) | NS | |
| Eosinophils (%), median (range) | 2 (0‐15) | 3 (0‐12) | 2 (0‐33) | 3 (0‐18) | 2 (0‐18) | NS | |
| Karyotype | Normal (%) | 97 | 77 | 69 | 40 | 86 | <.01 |
| Monosomy 7 (%) | 0 | 13 | 21 | 52 | 7 | ||
| Other aberration (%) | 3 | 10 | 10 | 8 | 7 |
Analysis of 407 children diagnosed between 1990 and 2018 registered to the European Working Group of MDS in Childhood (EWOG‐MDS).
AMC, absolute monocyte count; CM, costal margin; EP, erythropoiesis; MP, myelopoiesis.
Large spleens in pelvis set as 15 cm below costal margin.
Promyelocytes + myelocytes + metamyelocytes + normoblasts.
In ∼10% of JMML patients, a canonical alteration in PTPN11, NRAS, KRAS, CBL, or NF1 cannot be identified (Table 1). In a few of these cases, heterozygous somatic mutations in the RRAS gene have been described and shown to be initiating events.3-5 RRAS is a small guanosine triphosphatase (GTPase) that exhibits 50% to 60% homology to the RAS proteins and is associated with several diverse cellular processes.
Children with RRAS‐mutated JMML can have an atypical clinical course with rapid progression to acute myeloid leukemia (AML).3 Expanding the spectrum of RAS pathway mutations in JMML further, a case of a JMML patient with a presumed driver RRAS2 mutation has been noted.5
Patients with Noonan syndrome (NS) and PTPN11 germ line mutations can develop a self‐limiting myeloproliferative disorder (MPD) (see the following section). Although the hematological picture can be indistinguishable from JMML in some cases, the myeloproliferation is polyclonal in most instances, and thus, the disorder was not been included in the World Health Organization JMML category.1
NF1‐mutated JMML
Germ line mutations in NF1 are present in ∼10% of children with JMML (Table 1). Because the NF1 gene product, neurofibromin, is a GTPase-activating protein (GAP) and thus a negative modulator of RAS function, loss of heterozygosity with loss of the normal NF1 allele in leukemic cells is associated with RAS hyperactivity. In a study of 15 children with NF1 and JMML, two-thirds had loss of heterozygosity caused by segmental uniparental disomy of large parts of chromosome 17q, and in a minority of cases by somatic interstitial deletions, whereas one‐third had a compound‐heterozygous NF1‐inactivating mutation in leukemic cells.6 In most cases, the diagnosis of NF1 can be established clinically by the presence of ≥6 café au lait macules >0.5 cm in diameter. In addition, one-half of these children have a parent affected by NF1. However, in some children, JMML may be the first sign of NF1. Children with JMML and NF1 have a higher platelet count, a higher percentage of blasts in BM, and are more often diagnosed after the age of 5 years than JMML patients of other subtypes.2
PTPN11‐mutated JMML
PTPN11 is the most common mutated target in JMML with somatic PTPN11 mutations noted in up to 40% of JMML cases. The PTPN11 gene encodes for the cytoplasmic protein tyrosine phosphatase SHP‐2, which contains 2 tandem SH‐2 domains at the N terminus and a catalytic PTP domain at the C terminus. In its inactive state, PTPase activity is repressed by inhibition of the enzymatic cleft by the N‐terminal SH‐2 domain.7 All PTPN11 mutations are missense mutations in the N‐terminal SH‐2 (exon 3) or PTP interacting surfaces (exon 13) resulting in gain of function.8 As with NF1, JMML with PTPN11 mutation is a rapidly fatal disorder unless the patient undergoes hematopoietic stem cell transplantation (HSCT). In some HSCT series, patients with PTPN11 mutations had a significantly worse outcome with higher relapse rates when compared with patients of the other JMML genetic subtypes.9
NRAS‐mutated JMML
As with KRAS, NRAS mutations in JMML are typical cancer‐associated somatic heterozygous RAS mutations that introduce amino acid substitutions at positions Gly 12, Gly13, or Gln61 and lock RAS in the active guanosine triphosphate–bound state, thereby diminishing the intrinsic GTPase activity and/or by causing resistance to GAP.7 NRAS mutations at diagnosis are noted in 14% to 17% of cases (Table 1). Among the 5 genetic JMML subtypes, NRAS‐associated disease displays the greatest clinical diversity. Although a considerable percentage of patients transplanted for JMML with NRAS mutations relapse after HSCT,5,10 others survive in the absence of HSCT with persistence of NRAS mutation but slowly regressing disease.11 Clinically, these patients are well and show a normal or only slightly elevated HbF. Genetic and whole genome DNA methylation studies (see the following section) suggest that children with NRAS mutation and spontaneous regression have a low methylation profile without additional mutations.4,12‐14
KRAS‐mutated JMML
Children with somatic heterozygous KRAS mutations often have a clinically particular aggressive form of MPD. Median age is lower than that observed in PTPN11- or NF1-mutated cases (Table 2). Approximately one-half of the cases show monosomy 7.4 There is a puzzling overlap of clinical and laboratory features between RAS‐associated lymphoproliferative disease and JMML, which may represent different phenotypes of the same genetic disorder.15
CBL‐mutated JMML
Germ line mutations of the CBL gene cause CBL syndrome characterized by a high frequency of neurologic features/vasculitis, mild NS‐like features, and a high risk of JMML.16,17 CBL is a RING finger E3 ubiquitin ligase that regulates many proteins, including receptor tyrosine kinases. Germ line mutations in children with CBL‐mutated JMML are located throughout the linker and RING finger domain (intron 7, exons 8 and 9). Although most patients have 11q isodisomy in hematopoietic cells, a few heterozygous cases have been reported. Most children with JMML lack secondary genetic alterations (see the following section). Moreover, in the vast majority of children with CBL‐mutated JMML myeloproliferation is self‐limiting with splenomegaly decreasing over years.17
Distinguishing JMML from NS-associated myeloproliferative disease
Germ line mutations in literally every element of the RAS/mitogen activated kinase pathway have been discovered. These RASopathies, a class of developmental disorders, induce activation or dysregulation of the pathway, and thus share common clinical features such as facial dysmorphism, cardiac defects, reduced growth, and a variety of abnormalities in other organs.7
NS (MIM 163950) is the most genetically diverse and most common RASopathy occurring in 1 of 1000 to 2500 births.7 Heterozygous germ line mutations in PTPN11 account for about one-half of the cases; mutations in SOS1, RAF1, RIT1, KRAS, or other elements of the RAS pathway are noted in the remainder. Individuals with NS are predisposed to several hematological abnormalities, most commonly coagulation defects with thrombocytopenia, platelet dysfunction, and von Willebrand disease and factor deficiencies (reviewed in Briggs and Dickerman18 ). Although thrombocytopenia may be multifactorial, isolated severe thrombocytopenia with a decrease in megakaryocytes has been noted in some infants.19,20 Hepatosplenomegaly unexplained by cardiac failure is a common clinical finding in NS; it is more prominent in younger children and remits with age.
Excessive proliferation of myeloid (and in rare cases erythroid) precursors in NS was first been described about 20 years ago.21,22 In fact, ∼5% of neonates and infants with NS develop an MPD, which in its severe form clinically resembles JMML with hepatosplenomegaly, leukocytosis, and increase in blast percentage in PB and BM.23,24 The vast majority of these children with NS/MPD harbor germ line PTPN11 mutations predicted to result in a weaker gain‐of‐function effect than the somatic PTPN11 mutations in children with JMML.23,24
Underlying KRAS,25 NRAS,26 and RIT127 germ line mutations with MPD have been described in a few cases.
In most children with NS, MPD is benign and slowly regresses over months and years.23,24 However, a significant proportion of critically ill infants with heart disease, respiratory failure, MPD, and possibly other NS‐related succumb to their condition early in life.24 In these clinically compromised infants, a short course of mild cytoreductive therapy, such as treatment with 6‐mercaptopurine, can be helpful to ameliorate the deleterious effects of tissue invasion by abnormal myeloid cells.
The mechanism driving myeloid proliferation in NS/MPD remains unknown. Somatic mutations are conspicuously absent, as shown by French investigators in a large cohort of NS patients.24 Although polyclonality is assumed in most cases,28 results of X‐chromosome inactivation studies indicate clonal hematopoiesis in some instances,24,28 and there are rare cases of NS/MPD with secondary monosomy 7 who achieve spontaneous remission with persistence of monosomy 7 (Barbara de Moerloose, personal communication).29 Progression to AML has been reported in very few cases,21 thus, questioning recent recommendations for surveillance.30
Myeloproliferative neoplasia in early childhood mimicking JMML
Although receptor tyrosine kinase (RTK) translocations in hematological malignancies are relatively rare, single cases of MPD mimicking JMML in young children have been noted. Clinical evidence suggests that patients with these genetic abnormalities may benefit from RTK‐targeted inhibitors; thus, recognition of these translocations is crucial.
The presence of an ALK receptor tyrosine kinase rearrangement in an infant with atypical JMML was first described in 2010.31 In most of these rare instances, the ALK fusion partner is RAN‐binding protein 2 (RBP2, inv(2)(p23q13))14,31,32 ; fusion to the dynactin subunit 1 gene (DCTN1, t(2;12)(p23;q11)) has also been seen.14 Myeloid neoplasia with ALK rearrangement is associated with monosomy 7 across all age groups; loss of chromosome 7 occurs secondary to the splitting of the ALK gene.31,32 Although the blast percentage in ALK rearranged MPD/AML is generally rapidly progressing, single-agent ALK tyrosine kinase inhibitors can achieve hematological remission.14,32,33 The ROS1 receptor tyrosine kinase is structurally close to ALK, and a ROS1 fusion gene (TBL1XR1‐ROS1) was noted in a female adolescent with MPD.14
MPD with eosinophilia and constitutively activated platelet‐derived growth factor receptor α, platelet‐derived growth factor receptor β, or fibroblast growth factor receptor 1 are classed in a separate World Health Organization category. Occasionally, these myeloid neoplasms present in infancy with leukocytosis and organomegaly, and thus need to be differentiated from JMML with increased eosinophils.34,35
Apart from RTK fusion genes, dysregulated hematopoiesis with a transient myeloproliferation can be seen in an occasional infant with GATA2 deficiency (Marcin Wlodarski, personal communication).
Furthermore, a clinical presentation reminiscent of JMML with monocytosis, immature myeloid precursor on PB smear and moderate splenomegaly is noted in some patients with GATA2 deficiency and myeloid neoplasia.36
Last but not least, infant acute leukemia with KMT2A (MLL) rearrangement often presents with markedly increased liver and spleen size; thus, cases with low blast count need to be differentiated from JMML. In a 4-month-old boy with a sole translocation t(5;11)(q31;q23) the ρ GTPase activating protein 26 (ARHGAP26), also known as GTPase regulator associated with focal adhesion kinase, was fused to the KMT2 gene in hematopoietic cells. Following an initial JMML‐like clinical presentation, the child’s blast percentage rapidly increased.37
Nonmalignant disorders mimicking JMML
In patients with suspected JMML but absence of a RAS pathway lesion, nonmalignant disorders with a clinical and hematological picture mimicking JMML need to be excluded. Although presence of viral infections, Wiskott-Aldrich syndrome, or leukocyte adhesion deficiency is generally evident, radiographic imaging may be the key diagnostic method to clinically distinguish JMML from infantile malignant osteopetrosis with increased bone density and extramedullary hematopoiesis.38
Secondary molecular alterations in JMML
The somatic mutational landscape of patients with JMML has been extensively studied by Japanese, North American, and French investigators.4,5,14 In contrast to what has been observed in adult chronic myelomonocytic leukemia, the average number of mutations per sample is low (<0.5 mutations/Mb),4,5 confirming the paucity of mutational events required for JMML oncogenesis. In addition to the initiating RAS pathway mutation, secondary clonal abnormalities were detected in about one-half of the patients. The alterations occur in known oncogenes and tumor suppressor genes.
Approximately 10% to 15% of children are found to harbor RAS double mutants at diagnosis. Acquired NF1 haploinsufficiency in PTPN11‐mutated JMML is the most frequent of these changes, whereas secondary NRAS, KRAS, or CBL mutations are observed in all JMML subtypes with the exception of CBL‐mutated JMML.4,5,14 In line with previous case reports, duplication of oncogenic NRAS or KRAS from acquired uniparental disomy is associated with aggressive transformation of JMML.39
Components of the polycomb repressive complex 2 network like EZH2 and ASXL1, or other epigenetic modifiers like DNMT3A, are mutated in about 15% of JMML patients.4,5,40 EZH2 (7q31.2) point mutations are associated with monosomy 7, where they become hemizygous by loss of the wild-type allele.4,5
Although Japanese investigators uncovered JAK3 alterations in about 10% of their patients,14 the frequency of this alteration is lower in the French, Italian, and North American JMML cohorts.4,5,41 Recurrent mutations in the SKI domain of SETBP1 can be detected in 7% to 9% of JMML cases4,5,14,41 with droplet digital polymerase chain reaction identifying additional patients with small subclonal alterations.42 The spliceosome gene ZRSR2 is recurrently mutated in a few cases, pathogenic variants in SF3B1, U2AF1, or SRSF2 are, however, not found.4,5
In CBL‐mutated JMML, the only recurrent variant is copy‐neutral isodisomy at 11q23.3 where CBL is located, no other lesions are observed.4,5,14 Among the other JMML subtypes, frequency of secondary genetic alterations is similar, but patterns vary.4 KRAS‐mutated JMML displays monosomy 7 in about one-half of the cases, whereas alterations in the other subtypes were mostly point mutations.4
Epigenetic patterns in JMML
With the paucity of genetic lesions, and the failure of clinical and genetic markers to fully recapitulate heterogeneity in JMML, epigenetic patterns are of particular interest. Investigations of DNA methylation at the level of candidate gene promoters had identified DNA hypermethylation to be associated with poor clinical outcome.43 Most recently, 3 research groups performed an integrative analysis of genome‐wide DNA methylation profiles with mutational patterns, copy‐number changes, and gene expression in primary JMML samples.12‐14 Distinct methylation signatures correlated with clinical and genetic features and were highly predictive of relapse following HSCT. The high methylation group was characterized by somatic PTPN11 mutations and poor clinical outcome. The low methylation group was enriched for somatic NRAS and CBL mutations, and had a favorable prognosis. The intermediate methylation group showed enrichment for somatic KRAS mutations and monosomy 7.12
Risk assessment and therapy in JMML
High risk of early death and relapse following HSCT is clinically defined by age ≥2 years, platelet count ≤40 × 109/L, and an elevated HbF.2,44 Molecular risk factors in JMML include number of secondary clonal aberrations and DNA hypermethylation.4,5,14,43 Cross‐continental cooperation with harmonization of molecular approaches will facilitate the introduction of epigenetically defined risk stratification into future clinical trials.
Apart from most children with CBL‐mutated JMML, some NRAS‐mutated patients experience spontaneous regression of disease (see the previous section). These children are clinically well and show a low HbF. In the absence of rapidly available molecular biomarkers for risk determination, a careful watch-and-wait strategy may be indicated.
The majority of JMML patients require allogeneic HSCT for long‐term survival from leukemia.9,10 In fact, HSCT early in the course of disease improved the otherwise dismal prognosis of JMML patients significantly. Allogeneic HSCT, either from a histocompatible sibling or from an HLA‐matched/1‐antigen‐disparate unrelated donor, results in a disease‐free survival of 52%.44 Disease recurrence is the most important cause of failure, occurring with a cumulative incidence of 35%. Umbilical cord transplants are a suitable option for children lacking a HLA‐compatible relative.45 Standard preparative regimen consists of busulfan, cyclophosphamide and melphalan.44,46
A great variety of antineoplastic drugs has been applied pre‐HSCT, but none of these agents induced durable responses. There are several case reports of complete remissions achieved with the off‐label use of the hypomethylating agent azacytidine.47 Safety and efficacy of azacytidine is being evaluated in Europe in clinical trials for newly diagnosed (EudraCT Number 2014‐002388‐13) or relapsed (EudraCT number 2010‐022235‐10) JMML patients. To answer the question whether aberrant RAS signaling in JMML can be targeted by RAS pathway inhibition, the National Cancer Institute is currently sponsoring a clinical trial with the Children’s Oncology Group investigating the oral MEK inhibitor trametinib in relapsed and refractory JMML patients (NCT03190915).
Additional considerations
With the presence of canonical RAS pathway mutations in about 90% of patients, diagnosis of JMML is greatly facilitated. Because RAS pathway mutations may arise at a germ line or somatic level, analysis of nonhematopoietic tissue at presentation is strongly recommended for all patients. For timely diagnosis and decision making, we use DNA from hair follicles and buccal swabs (only useful when negative). With the high percentage of genetically defined disease and a better pathophysiological understanding, diagnostic criteria as published previously10 are no longer needed. In fact, this scoring system can be misleading and hinder careful exclusion of other rare MPD when canonical RAS pathway mutations are absent.14
Presence or absence of clonality is the major criteria to classify myeloproliferation in CBL syndrome as JMML, whereas the identical hematological and clinical picture in an individual with NS with germ line PTPN11 mutation is termed transient MPD. Because both JMML in CBL syndrome and transient MPD in PTPN11 mutated NS are generally self‐limiting disorders, the current classification is quite debatable. It needs to be recognized, however, that spontaneous regression in JMML is also found in nonsyndromic JMML patients. Furthermore, successful epigenetic interventions might decrease the number of patients in need for HSCT further. With their unique biology and underlying genetic defects, myeloproliferative diseases in young children will require careful clinical decision making.
Correspondence
Charlotte M. Niemeyer, Department of Pediatrics and Adolescent Medicine, University Children's Hospital, University of Freiburg, Mathildenstrasse 1, 79106 Freiburg, Germany; e-mail: charlotte.niemeyer@uniklinik-freiburg.de.
