
Abstract
Atypical hemolytic uremic syndrome (aHUS); hemolysis, elevated liver function tests, and low platelets syndrome; and transplant-associated thrombotic microangiopathy are related conditions, in that many patients harbor germline heterozygous mutations in genes that regulate the alternative pathway of complement (APC). Penetrance is variable because development of clinically significant disease appears to require supervention of a process such as inflammation. Complement activation on the endothelial surfaces leads to endothelial damage, platelet consumption, microthrombi, and a mechanical hemolytic anemia with schistocytes. Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal hematopoietic disease caused by expansion of a stem cell that harbors a somatic mutation in PIGA. PIGA mutant blood cells are deficient in the complement regulator proteins CD55 and CD59, making them susceptible to intravascular hemolysis due to a failure to regulate the APC on erythrocytes. Eculizumab is a monoclonal antibody that binds to C5 and inhibits terminal complement by interfering with the cleavage of C5 by the C5 convertases. The drug is approved by the US Food and Drug Administration for the treatment of aHUS and PNH; however, a new generation of complement inhibitors that block C5 and other components of the complement cascade is showing promise in preclinical and clinical trials.
Learning Objectives
Discuss mechanism(s) of complement-driven hemolytic anemias
Describe natural and pharmacologic inhibitors of the alternative pathway of complement
Complement activation and the alternative pathway
Aberrant regulation of the alternative pathway of complement (APC) (Figure 1) underlies the pathophysiology of thrombotic microangiopathies (TMAs), including atypical hemolytic uremic syndrome (aHUS), posttransplant TMAs, and hemolysis, elevated liver function tests, and low platelets (HELLP) syndrome.2 Deficiency of ≥1 complement regulatory protein on erythrocytes may also result in complement-driven hemolysis; examples include paroxysmal nocturnal hemoglobinuria (PNH) and germline CD59 deficiency.3,4 In addition to the canonical pathways (classical, lectin, and alternative), complement is activated by the coagulation and fibrinolytic pathways. For example, thrombin may function as a C3 and C5 convertase, and plasmin and kallikrein directly cleave C3 and its activation fragments; however, it is still not clear that this is biologically important in vivo.5 Regardless of how the complement cascade is initiated, it has been estimated that the APC accounts for >80% of complement activation products.6 This is because the alternative pathway C3 convertase (C3bBb) produces the C5 convertase, as well as cleaves more C3 molecules, leading to the rapid generation of C3b and resulting in a potent amplification loop.

Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Schematic diagram of the complement cascade. The 3 primary routes for activation of complement are the lectin pathway (LP), the classical pathway (CP), and the alternative pathway (AP). The LP and CP are activated when specific triggers are recognized by host pattern-recognition receptors. The AP is constitutively active. Initial activation through the LP or CP generates a shared C3 convertase (C4b·C2a). In the AP, C3b pairs with factor B (FB) to form the AP proconvertase (C3b·B), which is processed by factor D (FD) to form the AP C3 convertase (C3b·Bb·P). Both types of C3 convertases cleave C3 to generate C3a and C3b. C3a is a weak anaphylatoxin, a substance that promotes an inflammatory response. Nascent C3b binds covalently to cell surface proteins, forming the nidus of the C3 convertase. C3b that binds to the surface of a healthy host cell is quickly inactivated; C3b that attaches to the surface of a pathogen or altered host cell triggers a rapid amplification loop to generate more C3b, resulting in opsonization. C3b also complexes with the C3 convertases to form the C5 convertases (C4b·C2a·C3b and C3b·Bb·C3b·P). In the terminal complement cascade, C5 convertases cleave C5 into C5a (a strong anaphylatoxin) and C5b. C5b combines with C6-9 to form the membrane attack complex (MAC), also referred to as the terminal complement complex. Regulatory factors act at various stages of the cascade to control complement activation via their decay accelerating activity and/or cofactor activity. MASPs, mannose-binding lectin-associated serine proteases; MBL, mannose-binding lectin; PAMPs, pathogen-associated molecular patterns.
Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Schematic diagram of the complement cascade. The 3 primary routes for activation of complement are the lectin pathway (LP), the classical pathway (CP), and the alternative pathway (AP). The LP and CP are activated when specific triggers are recognized by host pattern-recognition receptors. The AP is constitutively active. Initial activation through the LP or CP generates a shared C3 convertase (C4b·C2a). In the AP, C3b pairs with factor B (FB) to form the AP proconvertase (C3b·B), which is processed by factor D (FD) to form the AP C3 convertase (C3b·Bb·P). Both types of C3 convertases cleave C3 to generate C3a and C3b. C3a is a weak anaphylatoxin, a substance that promotes an inflammatory response. Nascent C3b binds covalently to cell surface proteins, forming the nidus of the C3 convertase. C3b that binds to the surface of a healthy host cell is quickly inactivated; C3b that attaches to the surface of a pathogen or altered host cell triggers a rapid amplification loop to generate more C3b, resulting in opsonization. C3b also complexes with the C3 convertases to form the C5 convertases (C4b·C2a·C3b and C3b·Bb·C3b·P). In the terminal complement cascade, C5 convertases cleave C5 into C5a (a strong anaphylatoxin) and C5b. C5b combines with C6-9 to form the membrane attack complex (MAC), also referred to as the terminal complement complex. Regulatory factors act at various stages of the cascade to control complement activation via their decay accelerating activity and/or cofactor activity. MASPs, mannose-binding lectin-associated serine proteases; MBL, mannose-binding lectin; PAMPs, pathogen-associated molecular patterns.
Regulation of the alternative pathway
C3, factor B, factor D, and factor P (properdin) are the major components of the APC. The APC is activated by spontaneous hydrolysis of C3, which exposes a binding site for factor B. This “tickover” process maintains the APC in an activated state. Microbes are opsonized by complement and phagocytized by cells that have receptors that recognize components of the opsonization process. Microbes lack complement-regulatory proteins and are vulnerable to cell killing; humans possess a number of fluid-phase and cell surface proteins that abort complement activation. This is just 1 way in which the innate immune system distinguishes self from nonself. Once C3b binds to the cell, it forms a complex with factor B (C3bB). Factor D, a serine protease, then cleaves factor B into a soluble Ba and a Bb fragment. C3bBb is stabilized by factor P to form the APC C3 convertase C3bBbP. Some C3b is liberated by the convertase and binds to the complex itself, establishing the C5 convertase C3bBbC3bP. This new complex hydrolyzes the C5α-chain, liberating C5a and C5b (Figure 2). C5b binds to C6 and C7 and, in conjunction with C8 and several C9 molecules, forms the membrane attack complex (MAC). If C3b binds to human cells, serum proteins (factor H and factor I) and membrane proteins (complement receptor 1 [CR1], membrane cofactor protein [MCP/CD46], CD55, and CD59) restrict further formation and activation of the APC by degrading C3b or by interfering with the formation and stability of the C3 convertase. CD59 functions to block the MAC (Figure 2).
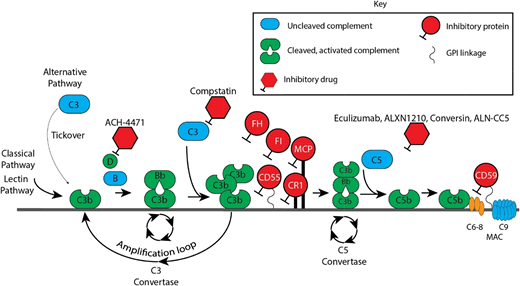
Complement cascade with inhibitors. CR1, membrane cofactor protein (MCP/CD46), factor I (FI), factor H (FH), and CD55 are the major proteins that inhibit the APC. Of these, FH is the most critical. FH is a fluid-phase protein that can bind to cells via its interaction with cell surface glycosaminoglycan and sialic acid residues. It inhibits the APC in the fluid phase and on the cell surface. It competes with factor B for C3b attachment, thereby limiting formation of the C3 convertase, and it accelerates decay of the C3 convertase. Deficiency or impaired function of FH leaves healthy host cells and tissues highly vulnerable to complement-mediated attack. Complement FI functions as a complement inhibitor in the presence of its cofactors (FH, MCP/CD46, and CR1), to cleave and inactivate C3b (in the alternative pathway) and C4b (in the classical pathway and lectin pathway), blocking C3 convertase formation.1 CD55 (also known as a decay-accelerating factor) is a cell surface glycosylphosphatidylinositol-anchored protein that inhibits the classical and alternative pathway C3 convertases and accelerates their decay. CD59 blocks terminal complement by preventing C9 from oligomerizing with the C5b-8 complex.
Complement cascade with inhibitors. CR1, membrane cofactor protein (MCP/CD46), factor I (FI), factor H (FH), and CD55 are the major proteins that inhibit the APC. Of these, FH is the most critical. FH is a fluid-phase protein that can bind to cells via its interaction with cell surface glycosaminoglycan and sialic acid residues. It inhibits the APC in the fluid phase and on the cell surface. It competes with factor B for C3b attachment, thereby limiting formation of the C3 convertase, and it accelerates decay of the C3 convertase. Deficiency or impaired function of FH leaves healthy host cells and tissues highly vulnerable to complement-mediated attack. Complement FI functions as a complement inhibitor in the presence of its cofactors (FH, MCP/CD46, and CR1), to cleave and inactivate C3b (in the alternative pathway) and C4b (in the classical pathway and lectin pathway), blocking C3 convertase formation.1 CD55 (also known as a decay-accelerating factor) is a cell surface glycosylphosphatidylinositol-anchored protein that inhibits the classical and alternative pathway C3 convertases and accelerates their decay. CD59 blocks terminal complement by preventing C9 from oligomerizing with the C5b-8 complex.
TMA-associated hemolytic anemias driven by complement
aHUS
aHUS is a complement-driven TMA characterized by hemolytic anemia, renal impairment, thrombocytopenia, and preserved ADAMTS13 function.2,7 aHUS results from complement-mediated damage (usually associated with a complement trigger, such as infection, surgery, pregnancy, cancer, or autoimmunity) to the microvascular endothelium, often in combination with germline defects in complement genes or autoantibodies against complement-regulatory proteins. Thus, penetrance is variable because development of clinically significant disease seems to require supervention of a process such as inflammation. Most mutations occur in genes that regulate the APC (factor H is the most common), but activating mutations in C3 and factor B are also found. Germline mutations that underlie aHUS are found in roughly 50% of patients, so absence of a germline variant does not exclude the diagnosis. The hemolytic anemia in aHUS is largely mechanical. Endothelial injury leads to platelet thrombi obstructing the microvascular and shearing of erythrocytes accounting for the presence of schistocytes, low haptoglobin, reticulocytosis, and elevated lactate dehydrogenase (LDH) levels. Endovascular injury predominantly affects glomeruli and arterioles in the kidney, but virtually any organ, including the brain, heart, lungs, gastrointestinal tract, and pancreas, may be involved. The diagnosis is suggested by the presence of a TMA, thrombocytopenia, and renal insufficiency in a patient with ADAMTS13 > 10% and a negative Shiga toxin. Laboratory assays, such as the modified Ham (mHam) test, can often distinguish between healthy thrombotic thrombocytopenic purpura (TTP) and aHUS by measuring intrinsic complement activation on a cell surface, but this assay is not clinically available.8 Prompt treatment with eculizumab, an anti-C5 monoclonal antibody that blocks formation of MAC, is indicated after a presumptive diagnosis has been established.9 The expeditious distinction between TTP and aHUS is clinically relevant, because plasma exchange does not arrest the complement-mediated organ damage occurring in aHUS, and early targeted therapy can preserve end-organ function. Improvement in the platelet count and LDH is usually seen within 48 to 72 hours of eculizumab administration. Kidney improvement may take several weeks or months. Eculizumab is administered IV every 7 days for the first 5 weeks and then every 2 weeks thereafter; however, the optimal duration of therapy is unclear. Although early reports suggested that long-term/indefinite therapy is required, more recent reports suggest that eculizumab may be safely discontinued in many aHUS patients.10-12 Before eculizumab is discontinued, the patient should be in complete remission (normal platelet counts, LDH, and renal function), and potential complement-activating “triggers” should be controlled. A discussion about the potential for relapse should take place with all patients before discontinuing eculizumab. Patients should be followed closely, especially for the first 4 to 6 months after discontinuing eculizumab, because this is the period during which there is the greatest risk for relapse. Two weeks after discontinuing eculizumab, the authors check the complete blood count with reticulocyte count, peripheral smear, serum creatinine, LDH, urine protein, history, and physical examination every week for 3 weeks, every 2 weeks for 8 weeks, every month for 2 months, and then every 3 months for 6 months. Patients who have received a solid organ transplant or those with an active autoimmune disease should probably remain on eculizumab indefinitely because of the risk for disease recurrence from such a nonreversible risk factor.
HELLP syndrome
HELLP syndrome is a severe variant of preeclampsia that affects up to 1% of all pregnancies13 and usually arises in the third trimester. Because disease signs and symptoms often resolve after delivery, iatrogenic preterm birth is a complication of management of HELLP. The pathogenesis is thought to be due to endothelial cell dysfunction; however, the mechanism is unclear. Therapeutic mainstays are supportive and include fetal monitoring, steroids for fetal lung maturity, magnesium for seizure prophylaxis, and management of hypertension.14 Fetal mortality approaches 30% when HELLP occurs early in the third trimester; maternal mortality may also approach 5% to 10%. Investigators have hypothesized that aHUS and HELLP may share a similar pathophysiology, because the clinical manifestations of hypertension, renal insufficiency, thrombocytopenia, elevated LDH, elevated aspartate aminotransferase, and even the presence of schistocytes are common to both disorders. Recent data from our laboratory using next-generation sequencing and functional complement assays in HELLP patients support this hypothesis.15,16 Similar to aHUS, rare germline variants (variant allele frequency < 1%) in genes regulating the APC (eg, C3, factor H, factor B, MCP) and/or activation of complement using the mHam test are found in >50% of patients with HELLP. The presence of a rare variant in a gene regulating the APC and an mHam test were highly specific for HELLP or aHUS (Figure 3). In contrast, no control patients (healthy third trimester pregnancy) were positive in both assays. These data suggest that a large subset of HELLP syndrome, similar to aHUS, is driven by an inability to regulate complement. The thrombocytopenia is consumptive, the hemolysis is mechanical, and the elevated “liver function tests” (LDH, bilirubin, and aspartate aminotransferase) are actually markers of intravascular hemolysis rather than intrinsic liver dysfunction. Germline mutations in genes that regulate the APC may predispose to HELLP. Complement levels normally rise after the second trimester of pregnancy and may serve as a trigger, along with other factors (eg, autoimmunity, infection), that contribute to vascular damage.17,18 Complement levels decrease following delivery, possibly explaining why the disease usually resolves postpartum. There are even case reports describing the use of eculizumab for treating HELLP, but this is not a US Food and Drug Administration–approved use.19
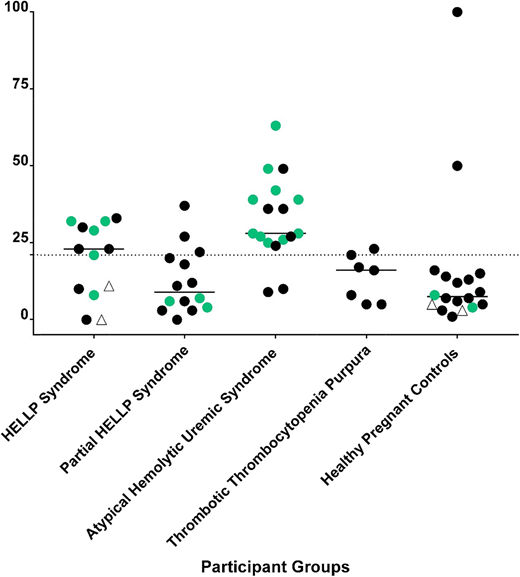
mHam test results and next-generation sequencing in TMAs. The mHam test is a cell-based in vitro viability assay that tests the ability of a PIGA mutant (CD55 and CD59–deficient) cell line to regulate complement. Each participant is represented by a symbol. A black circle indicates that no rare variants in APC-regulatory genes were detected; a green circle indicates that a rare variant was detected in an APC-regulatory gene, and an open triangle indicates that next-generation sequencing was not performed. A positive mHam test is defined as cell killing > 20.5% (dotted horizontal line) after exposure to patient serum. The median value of cell kill is depicted for each group. Patients with partial HELLP fit some, but not all, of the clinical criteria for HELLP.
mHam test results and next-generation sequencing in TMAs. The mHam test is a cell-based in vitro viability assay that tests the ability of a PIGA mutant (CD55 and CD59–deficient) cell line to regulate complement. Each participant is represented by a symbol. A black circle indicates that no rare variants in APC-regulatory genes were detected; a green circle indicates that a rare variant was detected in an APC-regulatory gene, and an open triangle indicates that next-generation sequencing was not performed. A positive mHam test is defined as cell killing > 20.5% (dotted horizontal line) after exposure to patient serum. The median value of cell kill is depicted for each group. Patients with partial HELLP fit some, but not all, of the clinical criteria for HELLP.
Transplant-associated TMA
Transplant-associated TMA (TA-TMA) is a rare life-threatening complication following blood and bone marrow transplantation that presents with thrombocytopenia, hemolytic anemia with schistocytes, renal dysfunction, hypertension, and sometimes altered mental status and/or seizures.20,21 The diagnosis can be challenging because patients are often anemic and thrombocytopenic related to the transplant-conditioning regimen for the first 4 to 6 weeks after bone marrow transplantation and may already be receiving red cell and platelet transfusions, obscuring the finding of schistocytes and platelet consumption. Furthermore, calcineurin inhibitors may lead to renal dysfunction and hypertension, and schistocytes may be the last finding to become clinically apparent. Withdrawal of calcineurin inhibitors will sometimes lead to resolution of the syndrome, but up to 70% of cases progress to severe end-organ damage and/or death. Plasma exchange is ineffective, but there are several successful reports of treating TA-TMA with complement inhibition.22 Similar to aHUS and HELLP, there is serological, cellular, and genetic evidence of complement driving the pathophysiology of the disease.23 Not all TA-TMA is complement driven; thus, identifying a reliable biomarker that can identify these cases will be helpful for future clinical trials of complement inhibition.24
Non-TMA anemias driven by complement
PNH
PNH is a clonal hematopoietic stem/progenitor cell disease that presents with hemolytic anemia, thrombosis, smooth muscle dystonias, and bone marrow failure.3 PNH is caused by clonal expansion of a stem/progenitor cell carrying a somatic PIGA mutation that results in an inability of cells to synthesize glycosylphosphatidylinositol (GPI). GPI serves as an anchor to attach dozens of proteins to the membrane of hematopoietic cells; thus, PIGA mutations lead to a deficiency of all GPI-anchored proteins, including the complement regulators CD55 and CD59. CD55 accelerates decay of the C3 and C5 convertases, and CD59 prevents formation of the MAC. PNH erythrocytes are particularly vulnerable to the absence of CD55 and CD59. Anucleated cells cannot repair membrane damage; thus, a single MAC forming on erythrocytes leads to hemolysis.25 PNH patients experience intravascular hemolysis because PNH red cells are defective in blocking formation of the MAC after C3b lands on the erythrocyte surface. Eculizumab binds to C5 and inhibits the C5 convertase (C3bBbC3b·P) from cleaving C5 into C5a and C5b; thus, it blocks complement upstream of CD59 and, thereby, decreases intravascular hemolysis by preventing formation of the MAC.26 However, because CD55 is upstream of C5, PNH patients on eculizumab accumulate C3 fragments on the PNH erythrocytes, and these patients have varying degrees of extravascular hemolysis.27 In the setting of strong complement activation (eg, infections, autoimmunity), dense C3b accumulation on PNH erythrocytes may result in a conformational change in C5 that disrupts the ability of eculizumab to block the cleavage of C5 by the C5 convertase and results in breakthrough hemolysis that resolves with resolution of the trigger.28
Eculizumab pros and cons
Eculizumab has changed the natural history of PNH.29-31 It blocks intravascular hemolysis, reduces or eliminates the need for blood transfusions, reduces the risk for thrombosis, and improves quality of life. Limitations include an increased risk for Neisseria infections (∼0.5% annually), cost, IV administration, and symptomatic extravascular hemolysis that occurs in up to 20% of PNH patients.32
Congenital CD59 deficiency
Rare patients with germline mutations affecting expression of CD59 have been described.4 In contrast to PNH, CD59 deficiency is on all cells, not just hematopoietic cells, and there is no GPI anchor deficiency; thus, CD55 and all other GPI-anchored proteins are present on the cell surface. These patients present with complement-mediated intravascular hemolysis and peripheral polyneuropathy. The neurologic symptoms are more prominent than hemolysis; however, intravascular hemolysis becomes more conspicuous following complement triggers, especially during infections. Administration of eculizumab has been shown to abrogate hemolysis in these patients and lead to neuronal regeneration.33
Novel drugs and targets for treating complement-mediated anemias
C5 inhibitors
There are more than a dozen novel complement inhibitors in preclinical or clinical development (Figure 2).34 Several compounds target C5 because terminal complement blockade is relatively safe and quite effective. Alexion Pharmaceuticals (New Haven, CT) has developed a new anti-C5 monoclonal antibody, ALXN1210 (ravulizumab), which is virtually identical to eculizumab but with a longer half-life; the drug is administered IV every 8 weeks after a brief induction period. An international randomized phase 3 trial of ravulizumab vs eculizumab has been completed, and results will be available this year. Another strategy to block C5 cleavage is being used by Alnylam Pharmaceuticals (Cambridge, MA), who developed a C5-specific N-acetylgalactosamine–conjugated small-interfering RNA duplex: ALN-CC5. This drug, in clinical trials, is administered subcutaneously weekly and efficiently silences C5 production in the liver. Coversin, developed by Akari Therapeutics (New York, NY), is derived from a small protein (16 kDa) isolated from the tick Ornithodoros moubata.35 The protein binds to C5 and blocks cleavage by C5 convertases. In vitro, the drug (daily, subcutaneous dosing) is effective in preventing hemolysis of PNH erythrocytes and was successfully used in a patient with TA-TMA carrying a C5 mutation associated with eculizumab resistance.36,37 Several other strategies to target C5 are in various stages of preclinical and clinical development. Although the results of these approaches are encouraging and exciting, it is unlikely that targeting C5 will solve the problem of extravascular hemolysis, because these therapies do not prevent the deposition of C3 fragments on the erythrocyte and resultant opsonization.
C3 inhibitors
In the complement activation cascade, C3 is upstream of C5; thus, blockage at the C3 level would theoretically prevent intravascular and extravascular hemolysis. C3 has a central critical role in all 3 complement pathways, which makes it a rational target, but it is also the most abundant complement factor in the serum (1.2 mg/mL), which makes it a pharmacologically challenging target. Compstatin is a 13-residue disulfide-bridged peptide that binds to human C3 and its active fragment C3b, thereby preventing C3 cleavage and the incorporation of C3b into the C3 and C5 convertases.38,39 A compstatin derivative from Apellis Pharmaceuticals (Crestwood, KY) is in clinical trials to treat PNH. Several other compstatin analogs are also being studied. Long-term safety will need to be assessed, because these compounds disable the classical, lectin, and alternative pathways.
Alternative pathway inhibitors
Factor D is the only enzyme in the blood that can activate factor B. The plasma concentration of factor D (1.8 ± 0.4 μg/mL) is the lowest of any complement protein, and this makes factor D a promising target for complement inhibition. Achillion Pharmaceuticals (New Haven, CT) has synthesized a small molecule (ACH-4471) that inhibits factor D. This compound can block PNH cell hemolysis, mitigate the accumulation of C3 fragments on the surface of PNH cells, as well as block the APC in in vitro models of aHUS.40 Human studies with oral ACH-4471 are underway. Drug development targeting other proteins of the alternative pathway (eg, factor B, factor H, and factor P) is also underway, and these are rational targets for treating PNH, aHUS, and possibly other hemolytic anemias. Clinical trials over the next 3 to 5 years will determine which, if any, of these new complement inhibitors is equivalent or superior to eculizumab in safety, efficacy, cost, and ease of administration.
Classical and lectin pathway inhibitors
Terminal complement activation occurs via 3 canonical pathways (classical, lectin, alternative), and targeting components of the classical and lectin pathways upstream of the alternative pathway may be therapeutically advantageous. Omeros Pharmaceuticals (Seattle, WA) has developed OMS721, a monoclonal antibody against mannan-binding lectin-associated serine protease 2. This IV administered agent is currently in phase 3 clinical trials for IgA nephropathy, TA-TMA, and aHUS. Although discussed elsewhere, cold agglutinin disease is also a complement-mediated hemolytic anemia driven by IgM and classical pathway activation. True North Therapeutics (San Francisco, CA) developed a monoclonal antibody (TNT-003) against C1s, and this agent was shown to be effective in ameliorating hemolysis in cold agglutinin disease.41
Correspondence
Robert A. Brodsky, Ross Research Building, Rm 1025, 720 Rutland Ave, Baltimore, MD 21205-2196; e-mail: brodsro@jhmi.edu.
