
Abstract
The advent of plasma exchange has dramatically changed the prognosis of acute thrombotic thrombocytopenic purpura (TTP). Recent insights into TTP pathogenesis have led to the development of novel therapies targeting pathogenic anti-ADAMTS13 antibody production, von Willebrand factor (VWF)–platelet interactions, and ADAMTS13 replacement. Retrospective and prospective studies have established the efficacy of rituximab as an adjunct to plasma exchange for patients with acute TTP, either upfront or for refractory disease. Relapse prevention is a major concern for survivors of acute TTP, and emerging data support the prophylactic use of rituximab in patients with persistent or recurrent ADAMTS13 deficiency in clinical remission. Capalcizumab, a nanobody directed against domain A1 of VWF that prevents the formation of VWF–platelet aggregates, recently completed phase 2 (TITAN) and 3 (HERCULES) trials with encouraging results. Compared with placebo, caplacizumab shortened the time to platelet recovery and may protect against microthrombotic tissue injury in the acute phase of TTP, though it does not modify the underlying immune response. Other promising therapies including plasma cell inhibitors (bortezomib), recombinant ADAMTS13, N-acetyl cysteine, and inhibitors of the VWF–glycoprotein Ib/IX interaction (anfibatide) are in development, and several of these agents are in prospective clinical studies to evaluate their efficacy and role in TTP. In the coming years, we are optimistic that novel therapies and international collaborative efforts will usher in even more effective, evidence-based approaches to address refractory acute TTP and relapse prevention.
Learning Objectives
Discuss current treatment of TTP
Examine the role of rituximab in acute TTP and during remission
Discuss caplacizumab and other novel targeted therapies for TTP
Introduction
Thrombotic thrombocytopenic purpura (TTP) is an acute, life-threatening, primary thrombotic microangiopathy characterized by microangiopathic hemolytic anemia, organ ischemia due to platelet-rich microthrombi, profound peripheral thrombocytopenia, and a severe deficiency of ADAMTS13, the von Willebrand factor (VWF) cleaving protease.1 ADAMTS13 deficiency in TTP is usually severe (<10% activity) and may be congenital due to mutations in the coding gene or, more commonly, acquired due to an autoantibody against the enzyme.1 The definitive demonstration in 1991 that plasma exchange (PEX) reduced mortality in TTP from >90% to <20%2,3 ushered in a new era of effective treatment of this historically fatal disorder. A deeper understanding of the mechanisms underlying TTP, including the central role of anti-ADAMTS13 antibodies in acquired TTP, has revolutionized the treatment of TTP, and immunosuppressive therapies play an important role as an adjunct to therapeutic PEX for refractory or relapsing TTP. Rituximab, the most successful of these immunomodulatory therapies,4 has found wide application in the treatment of refractory or relapsed TTP5,6 and is often used in the initial treatment of TTP and as preemptive therapy to prevent relapse,7 although the latter uses remain controversial.
Finally, VWF, the protein that crosslinks platelets to form the platelet-rich microthrombi in TTP, has emerged as an important target in TTP. Caplacizumab, a nanobody directed against the platelet-binding domain of VWF,8,9 has demonstrated efficacy in randomized trials and is expected to be available in the near future. Figure 1 highlights important milestones in TTP therapy. Despite these advancements, ∼15% of patients still die of TTP, and several questions remain unanswered. What is the best approach to TTP that responds to PEX poorly, or not at all? Should we use rituximab routinely in the initial treatment of TTP or in remission to prevent relapse? What is the optimal timing, dose, and schedule of rituximab administration? Finally, how should we integrate newer therapies into current, generally effective paradigms of TTP management?
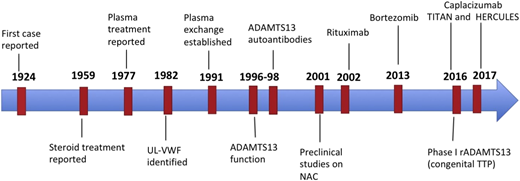
Milestones in TTP therapy. The major developments since TTP was first described were the discovery that PEX led to recovery in the majority of patients; the identification of the role of ultra-large VWF multimers, ADAMTS13, and anti- ADAMTS13 antibodies in the pathogenesis of TTP; and the use of rituximab for acute and relapsing TTP. The last decade has seen the development of multiple promising novel targeted therapies.
Milestones in TTP therapy. The major developments since TTP was first described were the discovery that PEX led to recovery in the majority of patients; the identification of the role of ultra-large VWF multimers, ADAMTS13, and anti- ADAMTS13 antibodies in the pathogenesis of TTP; and the use of rituximab for acute and relapsing TTP. The last decade has seen the development of multiple promising novel targeted therapies.
Current treatment of acquired TTP
Timely diagnosis of TTP is crucial, because TTP responds well to PEX but has mortality in excess of 90% if left untreated. Because of its varied and often nonspecific presentation, a high index of suspicion is critical. The classic pentad of TTP, including microangiopathic hemolytic anemia, thrombocytopenia, fever, renal failure, and neurologic abnormalities, occurs in <5% to 10% of cases.10 In fact, microangiopathic hemolysis characterized by red cell fragmentation on the blood smear and thrombocytopenia are the only consistent features and, in the absence of other obvious causes, should prompt immediate treatment of presumed TTP. ADAMTS13 activity <10% in the setting of an acute thrombotic microangiopathy confirms the diagnosis of TTP, but this specialized assay is not immediately available at all institutions. TTP is a medical emergency, and prompt initiation of PEX should not wait on laboratory confirmation of the diagnosis. While making a clinical distinction between TTP and other thrombotic microangiopathies such as atypical hemolytic syndrome, scleroderma renal crisis, HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome, and malignant hypertension can be challenging early in the course; these should be considered in the appropriate clinical setting, particularly when renal involvement is prominent. Clinical prediction rules such as the PLASMIC score can facilitate early treatment decisions by identifying patients who would benefit from PEX. The PLASMIC score allots 1 point each for platelet count, hemolysis variable, absence of cancer, no solid organ or stem cell transplantation, international normalized ratio <1.5, and serum creatinine <2 mg/dL and categorizes patients as low (score 0-4), intermediate (score 5), and high (score 6 or 7) risk for TTP.11 Figure 2 outlines our approach to managing acquired TTP.
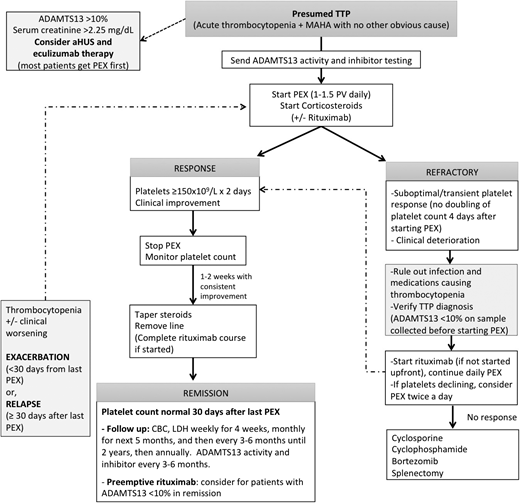
Approach to the management of TTP. For patients presenting with microangiopathic hemolysis without another obvious cause, start PEX promptly for a presumed diagnosis of TTP. ADAMTS13 activity and inhibitor testing should be sent before starting PEX, which can affect the results. We start corticosteroids as an adjunct to PEX in all patients and rituximab for patients who have severe symptoms (neurologic or cardiac involvement) or a suboptimal response for PEX (no doubling of platelet count in 4-5 days). PEX can be stopped or tapered once platelet count has been normal (>150 × 109/L) on 2 occasions at least 24 hours apart, and steroids can be tapered. For patients with refractory TTP or worsening thrombocytopenia after an initial response, we recommend verifying that ADAMTS13 activity is <10% before starting PEX (if available), ruling out infections or medications that can contribute to thrombocytopenia and then considering therapy with rituximab (if not already started). For TTP that is refractory to these measures, other immunosuppressants such as cyclosporine, cyclophosphamide, and bortezomib, or even splenectomy, may be considered. aHUS, atypical hemolytic uremic syndrome; CBC, complete blood count; LDH, lactate dehydrogenase; MAHA, microangiopathic hemolytic anemia; PV, plasma volume.
Approach to the management of TTP. For patients presenting with microangiopathic hemolysis without another obvious cause, start PEX promptly for a presumed diagnosis of TTP. ADAMTS13 activity and inhibitor testing should be sent before starting PEX, which can affect the results. We start corticosteroids as an adjunct to PEX in all patients and rituximab for patients who have severe symptoms (neurologic or cardiac involvement) or a suboptimal response for PEX (no doubling of platelet count in 4-5 days). PEX can be stopped or tapered once platelet count has been normal (>150 × 109/L) on 2 occasions at least 24 hours apart, and steroids can be tapered. For patients with refractory TTP or worsening thrombocytopenia after an initial response, we recommend verifying that ADAMTS13 activity is <10% before starting PEX (if available), ruling out infections or medications that can contribute to thrombocytopenia and then considering therapy with rituximab (if not already started). For TTP that is refractory to these measures, other immunosuppressants such as cyclosporine, cyclophosphamide, and bortezomib, or even splenectomy, may be considered. aHUS, atypical hemolytic uremic syndrome; CBC, complete blood count; LDH, lactate dehydrogenase; MAHA, microangiopathic hemolytic anemia; PV, plasma volume.
PEX
Therapeutic PEX is the mainstay of management of acute TTP and has improved survival rates from <20% to >80%.2,3,12 PEX replenishes ADAMTS13 activity and removes anti-ADAMTS13 antibodies, ADAMTS13 immune complexes, and ultra-large VWF multimers. The effectiveness of PEX in TTP was conclusively established by a prospective trial from the Canadian Apheresis Group, which randomized patients with TTP to either PEX or plasma infusion and reported lower early mortality with PEX than plasma infusion (3.9% vs 15.7%, P = .035) and improved survival at 6 months (78% vs 63%, P = .002).2 A large observational study of 108 patients (of whom 78 received PEX) also reported similar outcomes, with 91% of patients surviving the acute episode.3 The optimal PEX regimen has not been established. Our practice, based on the Canadian apheresis trial and the British Committee on Standards in Haematology guidelines,2,12 is to start with PEX at 1.5 PV exchanges for the first 3 days and 1 PV thereafter; however, others recommend 1 PV from the beginning13 ; the superiority of one strategy over the other has not been demonstrated. PEX is continued until the platelet count normalizes (≥150 × 109/L for at least 2 consecutive days). Many groups will stop PEX after achieving a stable response, while others recommend tapering the frequency of PEX in an attempt to prevent exacerbations (recurrent TTP within 30 days of the last PEX).13,14 Evidence supporting this practice is limited, and exacerbation and relapse (recurrent TTP ≥30 days after the last PEX) are expected to be less common in patients treated with rituximab (and potentially caplacizumab in the future). Different plasma preparations, such as fresh frozen plasma, virally inactivated solvent/detergent plasma, and psoralen-derivative plasma, appear to be equally effective in treating TTP.15 Case reports suggest that cryo-poor plasma, which lacks ultra-large VWF multimers, is effective in the treatment of some patients with refractory TTP, but other studies failed to demonstrate any advantage over fresh frozen plasma in upfront therapy.16,17
Immunomodulatory therapies
Corticosteroids
Most patients with acquired TTP receive corticosteroids in addition to PEX. This practice is supported by the autoimmune nature of the disease and an early report in which 30 of 54 patients with less severe TTP responded to steroid therapy alone in 48 to 72 hours.3 A small multicenter randomized study suggested that high-dose methylprednisolone (10 mg/kg per day for 3 days, followed by 2.5 mg/kg per day) might be superior to standard-dose methylprednisolone (1 mg/kg per day).18 A recent study prospectively randomized 26 patients to either cyclosporine or prednisone.19 Although there was no significant difference in the exacerbation rate, suppression of anti-ADAMTS13 antibodies and improvement in ADAMTS13 activity in the first month after stopping PEX were significantly better in prednisone-treated subjects.19 Taken together, these findings support the benefit of corticosteroids in acute TTP, and we start either prednisone 1 mg/kg per day or methylprednisolone 1 g per day for 3 days (followed by prednisone 1 mg/kg per day) as an adjunct to plasma therapy and taper the dose after achieving a stable response.12
Rituximab
Rituximab, a chimeric monoclonal antibody directed against CD20 on mature B cells, was approved by the US Food and Drug Administration for the treatment of non-Hodgkin lymphomas in 1997 but has since found wide application in the treatment of autoimmune diseases, including TTP (in which it is used off label). Its efficacy in TTP is through B-cell depletion, which suppresses the production of anti-ADAMTS13 autoantibodies and raises ADAMTS13 activity.14,20 Conversely, relapses of TTP after rituximab treatment correlate with a decrease in anti-ADAMTS13 activity.7 Rituximab was initially used in patients refractory to PEX and corticosteroids, but it is now increasingly used in the initial treatment of patients presenting with TTP based on observational studies indicating that rituximab may hasten recovery and reduce exacerbation and relapse rates.4,21 Table 1 summarizes selected studies (with at least 20 patients each) evaluating rituximab therapy in TTP.
Summary of studies (including ≥20 patients) evaluating rituximab therapy for TTP
| Reference | Study design | No. of patients | Dose and schedule | CR rate (%) | Time to CR (days) | Relapse rate | Comments |
|---|---|---|---|---|---|---|---|
| Upfront for the initial treatment of TTP | |||||||
| Scully et al7 | Prospective multicenter phase 2 (matched historical control group) | 40 (40 historical controls matched for age, sex, ethnicity, and number of relapses) | 375 mg/m2 per wk, 4 doses (up to 8 doses for persistent anti-ADAMTS13 antibodies) | 82.5 | 12 | 10% vs 57% in historical controls; median time to relapse, 27 (17-31) months for rituximab group and 18 (3-60) months for controls | Median duration of hospitalization was reduced by 7 d in patients who did not need intensive care admission; possible selection bias with controls who had a slightly higher relapse rate (57%) than shown in other cohorts; ADAMTS13 activity <10% was not confirmed in all patients |
| Westwood et al24 | Single-center retrospective case series | 100 (86 rituximab naive and 14 pretreated) | 375 mg/m2 per dose weekly or twice a week, 4 doses (up to 8 infusions for patients with persistent anti-ADAMTS13 antibodies) | 95, rituximab naive; 88, prior rituximab | 14 (4-52) | 13.4% Median time to relapse 24 (4-49) months | Earlier administration of rituximab (≤3 d or >3 d from admission) was associated with fewer TPEs for remission (16 vs 24, P = 0.03), and shorter median length of stay (16 vs 23 d, P = 0.01); similar response to weekly or twice-weekly dosing |
| Treatment of refractory or relapsed TTP | |||||||
| Scully et al20 | Case series | 25 (14 refractory and 11 relapsed) | 375 mg/m2 per wk, 4 doses | 100 | 11 (7-12) | No relapses at median follow up of 10 mo (range, 1-33 mo) | 21/25 patients had normal ADAMTS13 activity at 3 mo and 23/25 had no detectable inhibitor |
| Froissart et al14 | Prospective multicenter phase 2 | 21 (18 first episode, 3 relapsed) with matched historical controls | 375 mg/m2 per dose on days 1, 3, 7, and 14 after determination of refractory TTP | — | — | 40% (2 relapses at 6 mo and 51 mo) | ADAMTS13 activity ranging from 15% to 75% with disappearance of inhibitors was achieved after 3 mo in all patients |
| Clark et al6 | Prospective multicenter single arm phase 2 | 40 (20 refractory, 20 relapsed) | 375 mg/m2 per wk, 4 doses | Refractory: 63.6 at 8 wk, 88.9 at 1 y; relapsed: 90% at 8 wk, 100 at 1 y | — | — | — |
| Page et al22 | Single-center cohort | 16 (14 refractory) treated with rituximab and 21 not treated | 375 mg/m2 per wk, 4 doses | — | — | 12.5% in the rituximab treated group vs 42.8% in the non–rituximab-treated group | — |
| Treatment of persistent anti-ADAMTS13 antibodies in remission | |||||||
| Westwood et al24 | Single-center retrospective case series | 15 patients (21 episodes). | 375 mg/m2 per wk, 4 doses (n = 13) or 100 mg/m2 per wk, 4 doses (n = 2) | — | — | 1 relapse over median follow-up of 23 mo (range, 1-89 mo) | ADAMTS13 activity normalized at 3 mo in 16/17 episodes |
| Hie et al29 | Cross-sectional study | 30 treated with rituximab and 18 not treated with rituximab in remission | 375 mg/m2 per wk, 4 doses | — | — | 0 episodes/y and 0.5 episodes/y with and without preemptive rituximab, respectively | — |
| Westwood et al28 | Retrospective multicenter case series | 45 (76 episodes) | 375 mg/m2 per wk, 4 doses (n = 24);200 mg per wk, 4 doses (n = 19); and 500 mg per wk, 4 doses (n = 17) | — | — | 3.9% | All relapses occurred in the reduced-dose (200 mg/wk) group, which also had a high rate of retreatment |
| Reference | Study design | No. of patients | Dose and schedule | CR rate (%) | Time to CR (days) | Relapse rate | Comments |
|---|---|---|---|---|---|---|---|
| Upfront for the initial treatment of TTP | |||||||
| Scully et al7 | Prospective multicenter phase 2 (matched historical control group) | 40 (40 historical controls matched for age, sex, ethnicity, and number of relapses) | 375 mg/m2 per wk, 4 doses (up to 8 doses for persistent anti-ADAMTS13 antibodies) | 82.5 | 12 | 10% vs 57% in historical controls; median time to relapse, 27 (17-31) months for rituximab group and 18 (3-60) months for controls | Median duration of hospitalization was reduced by 7 d in patients who did not need intensive care admission; possible selection bias with controls who had a slightly higher relapse rate (57%) than shown in other cohorts; ADAMTS13 activity <10% was not confirmed in all patients |
| Westwood et al24 | Single-center retrospective case series | 100 (86 rituximab naive and 14 pretreated) | 375 mg/m2 per dose weekly or twice a week, 4 doses (up to 8 infusions for patients with persistent anti-ADAMTS13 antibodies) | 95, rituximab naive; 88, prior rituximab | 14 (4-52) | 13.4% Median time to relapse 24 (4-49) months | Earlier administration of rituximab (≤3 d or >3 d from admission) was associated with fewer TPEs for remission (16 vs 24, P = 0.03), and shorter median length of stay (16 vs 23 d, P = 0.01); similar response to weekly or twice-weekly dosing |
| Treatment of refractory or relapsed TTP | |||||||
| Scully et al20 | Case series | 25 (14 refractory and 11 relapsed) | 375 mg/m2 per wk, 4 doses | 100 | 11 (7-12) | No relapses at median follow up of 10 mo (range, 1-33 mo) | 21/25 patients had normal ADAMTS13 activity at 3 mo and 23/25 had no detectable inhibitor |
| Froissart et al14 | Prospective multicenter phase 2 | 21 (18 first episode, 3 relapsed) with matched historical controls | 375 mg/m2 per dose on days 1, 3, 7, and 14 after determination of refractory TTP | — | — | 40% (2 relapses at 6 mo and 51 mo) | ADAMTS13 activity ranging from 15% to 75% with disappearance of inhibitors was achieved after 3 mo in all patients |
| Clark et al6 | Prospective multicenter single arm phase 2 | 40 (20 refractory, 20 relapsed) | 375 mg/m2 per wk, 4 doses | Refractory: 63.6 at 8 wk, 88.9 at 1 y; relapsed: 90% at 8 wk, 100 at 1 y | — | — | — |
| Page et al22 | Single-center cohort | 16 (14 refractory) treated with rituximab and 21 not treated | 375 mg/m2 per wk, 4 doses | — | — | 12.5% in the rituximab treated group vs 42.8% in the non–rituximab-treated group | — |
| Treatment of persistent anti-ADAMTS13 antibodies in remission | |||||||
| Westwood et al24 | Single-center retrospective case series | 15 patients (21 episodes). | 375 mg/m2 per wk, 4 doses (n = 13) or 100 mg/m2 per wk, 4 doses (n = 2) | — | — | 1 relapse over median follow-up of 23 mo (range, 1-89 mo) | ADAMTS13 activity normalized at 3 mo in 16/17 episodes |
| Hie et al29 | Cross-sectional study | 30 treated with rituximab and 18 not treated with rituximab in remission | 375 mg/m2 per wk, 4 doses | — | — | 0 episodes/y and 0.5 episodes/y with and without preemptive rituximab, respectively | — |
| Westwood et al28 | Retrospective multicenter case series | 45 (76 episodes) | 375 mg/m2 per wk, 4 doses (n = 24);200 mg per wk, 4 doses (n = 19); and 500 mg per wk, 4 doses (n = 17) | — | — | 3.9% | All relapses occurred in the reduced-dose (200 mg/wk) group, which also had a high rate of retreatment |
CR, complete response.
Rituximab for refractory TTP.
Early case reports and series employing rituximab along with continued daily PEX in patients with TTP who had a suboptimal response to standard PEX therapy reported encouraging results; however, rituximab was often administered along with other salvage therapies, and it is difficult to clearly attribute response to rituximab. In 2007, the UK group reported on 25 patients treated with rituximab for refractory (n = 14) or recurrent (n = 11) TTP.20 Stable platelet count recovery occurred in all patients in 7 to 21 days, and no relapses had occurred at a median follow-up of 19 months. ADAMTS13 activity also normalized in all patients by 3 months, and 23 patients had no detectable inhibitor. The French reference center for thrombotic microangiopathies compared the outcomes of 21 patients with refractory TTP treated with rituximab with 53 historical controls and reported that the rituximab group had a higher rate of recovery (100% vs 78%) and a lower relapse rate (0% vs 9.4%) at 1 year.14 Data from the Oklahoma TTP registry also report lower relapse rates in patients (mostly with refractory TTP) who received rituximab during the acute episode compared with those that did not receive rituximab (12.5% vs 42.8%).22
The optimal dose and schedule of rituximab therapy for TTP is still not established. The most common approach has been to use 375 mg/m2 weekly or twice a week for 4 to 8 doses, based on the doses used for lymphoma. Recent case series suggest that lower doses of rituximab may have comparable efficacy in acute TTP.23 A single-arm phase 2 trial enrolled 24 patients with refractory TTP and administered 2 doses of rituximab 375 mg/m2 on days 1 and 4 (after determination of refractory disease) with a third dose on day 15 for patients who still had detectable B lymphocytes.5 Compared with a historical control group (N = 22) treated with 4 doses of rituximab at 375 mg/m2 per week, the study group had comparable rates of remission at 30 days (95% vs 90%), total number of PEXs, and time to remission.5 An ongoing study is evaluating the efficacy of low-dose rituximab (100 mg per week) in acute TTP (NCT01554514).
Rituximab in the initial treatment of TTP.
In 2011, Scully et al reported a phase 2 trial investigating upfront rituximab with daily PEX in 40 patients with acute TTP.7 Rituximab 375 mg/m2 for 4 weekly doses was started within 72 hours of TTP diagnosis, and outcomes were compared with 40 historical controls matched for age, sex, and ethnicity. The median time to complete response was 12 days. The median duration of hospitalization was shortened by 7 days when patients that needed intensive care admission were excluded from the analysis. Most strikingly, the relapse rate was reduced to 10% compared with 57% in the historical controls.7 A subsequent study from the UK group reported that upfront rituximab was associated with fewer PEXs for remission and a shorter time to remission and discharge.24 These results encourage the use of rituximab at diagnosis of TTP; however, none of these studies were randomized, and a proportion of patients with TTP will recover without future relapses without rituximab therapy, so this approach could lead to overtreatment in these individuals. Moreover, it is unclear whether administering rituximab in the acute phase has superior outcomes compared with prophylactic rituximab in remission for patients with persistent ADAMTS13 deficiency or declining ADAMTS13. Our practice is to introduce rituximab early for patients who have a suboptimal response to PEX (no doubling of platelet count in 4-5 days) or severe symptoms at presentation (neurologic deficits or evidence of cardiac involvement).
When rituximab is used in the setting of ongoing daily PEX for acute TTP, whether upfront or for refractory/relapsing TTP, there is often a concern regarding removal of drug during PEX. Early studies (dosing rituximab at 375 mg/m2 per dose and avoiding PEX for 24 hours after the rituximab dose) established that PEX removes 60% to 70% of rituximab; however, rapid depletion of CD19-positive B cells and suppression of anti-ADAMTS13 antibody are still achieved.25 In their phase 2 study of rituximab for upfront treatment of TTP, Scully et al administered rituximab with a minimum of 4 hours between the infusion and further PEX therapy and still achieved suppression of ADAMTS13 antibody and recovery of ADAMTS13 activity.7 Our practice is to administer rituximab immediately after PEX and perform the next PEX after 24 hours.
Prophylactic rituximab in remission: “watch and wait” vs active treatment
Relapses occur in 40% of patients who recover after an episode of TTP and are associated with significant mortality and morbidity.1 Decreased ADAMTS13 activity in remission is an important risk factor for relapse. Investigators from the Ohio State University collected 157 serial samples from 24 patients at 3-month intervals during remission and showed that lower ADAMTS13 activity was associated with a higher rate of relapse in the subsequent 3 months (Figure 3).26 Peyvandi et al also showed that ADAMTS13 activity <10% at least 30 days after the last PEX increased the odds of relapse (odds ratio, 2.9; 95% confidence interval, 1.3-6.8), which was increased even further when both ADAMTS13 activity <10% and anti-ADAMTS13 antibodies were present (odds ratio, 3.6; 95% confidence interval, 1.4-9.0).27 Westwood et al reported that ADAMTS13 activity normalized in 15 patients treated with preemptive rituximab in remission and a single relapse over a median follow-up of 23 months (1-89 months).24 In another study, the UK group evaluated three different doses of rituximab (375 mg/m2 per dose, 200 mg per dose, or 500 mg per dose, each administered for 4 treatments) to prevent TTP relapse and reported that normalization of ADAMTS13 activity occurred in 79.8%; only 3 relapses (all in the reduced-dose group [200 mg per dose]) occurred.28 A cross-sectional French study reported that the relapse rate over 12 year follow-up of 48 patients with ADAMTS13 deficiency in remission was 0 episodes per year among 30 patients treated with preemptive rituximab compared with 0.57 episodes per year among 18 rituximab-untreated patients.29 Despite the mounting evidence supporting preemptive rituximab to prevent relapse, the optimal frequency of monitoring during clinical remission and the ADAMTS13 activity threshold below which rituximab should be offered have not been established. Our practice is to offer prophylactic rituximab 375 mg/m2 per week for 4 doses to patients with severe ADAMTS13 deficiency in remission (ADAMTS13 activity is <10% to 20%). If ADAMTS13 activity does not recover after rituximab therapy, we do not offer treatments other than rituximab in the absence of clinical evidence of a relapse. We monitor ADAMTS13 activity every 3 months for the first year, every 3 to 6 months until 2 years from diagnosis, and annually thereafter. However, the monitoring plan is individualized to some extent, and patients with frequent relapses may benefit from closer monitoring even several years after their initial diagnosis.
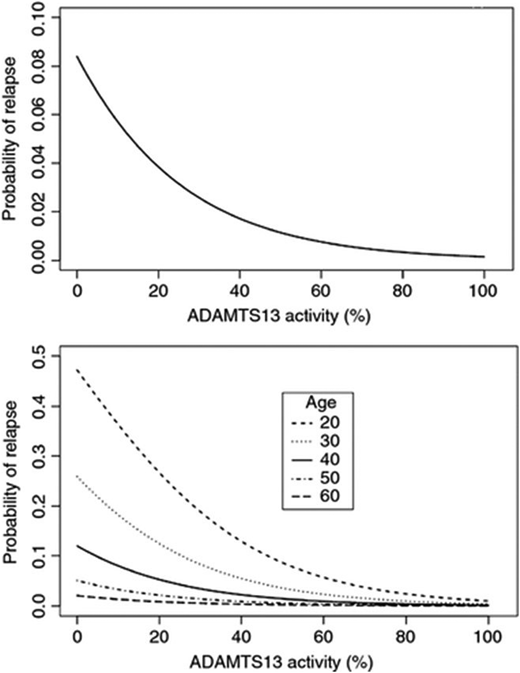
ADAMTS13 activity during remission and risk of relapse. Serial samples were collected at 3-month intervals during clinical remission in patients with acquired TTP. Logistic regression analysis showed that lower ADAMTS13 activity was associated with a higher probability of relapse over the following 3 months. This relationship held true over different age groups, though younger age was also associated with a higher risk of relapse. Adapted from Jin et al,26 with permission.
ADAMTS13 activity during remission and risk of relapse. Serial samples were collected at 3-month intervals during clinical remission in patients with acquired TTP. Logistic regression analysis showed that lower ADAMTS13 activity was associated with a higher probability of relapse over the following 3 months. This relationship held true over different age groups, though younger age was also associated with a higher risk of relapse. Adapted from Jin et al,26 with permission.
Other immunosuppressant therapies
Immunomodulatory agents such as vincristine and cyclosporine were popular as salvage therapy for patients with a poor response to PEX in the pre-rituximab era. Vincristine, in particular, was historically used to treat refractory or relapsing TTP with reported response rates ranging from 50% to 80%,30 an estimate that may be inflated by publication bias, as poor outcomes are less likely to be reported. Cyclosporine has been used successfully to treat refractory TTP and as an adjunct to PEX in the frontline therapy of TTP, an effect that correlates with suppression of ADAMTS13 antibodies and recovery of ADAMTS13 activity.31 However, a recent randomized trial did not show any benefit over prednisone in terms of exacerbation rates or suppressing anti-ADAMTS13 activity.19 Cyclophosphamide may also be effective in raising platelet counts in TTP.32 These therapies are now usually reserved for patients refractory to rituximab or in combination with rituximab and other therapies for patients refractory to PEX and steroids.
Splenectomy for refractory or relapsing TTP
Splenectomy has been used to treat patients with refractory and relapsing TTP as well as prophylactic therapy in patients with multiple relapses.32,33 Outcomes have been variable, with some series reporting minimal benefit and a complicated postsplenectomy course,3,32 and others reporting quick remissions and low relapse rates.33 A systematic review including 18 studies reported a response rate of 92% in 74 patients who underwent splenectomy for refractory TTP. Postsplenectomy relapse rate was reduced to 17% among 87 patients who underwent splenectomy for relapsing TTP.34 Splenectomy is a potentially effective therapy in TTP that works by eliminating a source of pathologic antibody production35 ; however, it has fallen out of favor since the availability of more effective pharmacologic immunomodulation.
Novel targeted therapies for acquired TTP
Novel therapies for acquired TTP target VWF–platelet interactions, anti-ADAMTS13 antibody production, and ADAMTS13 replacement therapy (Table 2). Targeting VWF has been particularly successful, because inhibiting the interaction between VWF and platelets can hasten platelet recovery and potentially prevent or minimize ischemic complications.
Novel targeted therapies for TTP
| Therapy | Mechanism/target | Supporting evidence |
|---|---|---|
| Rituximab | Suppress anti-ADAMTS13 antibody by depleting CD20-positive B cells | Retrospective and prospective studies show benefit in acute TTP and for relapse prevention (Table 1) |
| Caplacizumab | Nanobody directed toward the A1 domain of VWF inhibits VWF’s interaction with platelet GP1b-IX-V receptors | Phase 2 and 3 trials demonstrated reduced time to platelet normalization, lower exacerbation rates, and reduction in mean PEX procedures and hospital length of stay compared with placebo |
| NAC | Cleaves VWF multimers by reducing disulfide linkages | Reduction in VWF multimer size and VWF-dependent platelet aggregation in preclinical models |
| Case reports of successful use in refractory TTP | ||
| Bortezomib | Reduction in anti-ADAMSTS13 antibody production through apoptotic destruction of plasma cells | Case reports and series of successful use in patients refractory to multiple lines of therapy |
| Recombinant ADAMTS13 | Neutralization of anti-ADAMSTS13 antibodies and restoration of ADAMSTS13 activity | Phase 1 trial demonstrated safety in congenital TTP; phase 3 trial planned |
| Anfibatide | Reduces platelet–VWF interaction through antagonism of the platelet GP1b receptor | Inhibited platelet–VWF aggregate formation in in vitro studies and resolved thrombocytopenia in a mouse model of TTP |
| Therapy | Mechanism/target | Supporting evidence |
|---|---|---|
| Rituximab | Suppress anti-ADAMTS13 antibody by depleting CD20-positive B cells | Retrospective and prospective studies show benefit in acute TTP and for relapse prevention (Table 1) |
| Caplacizumab | Nanobody directed toward the A1 domain of VWF inhibits VWF’s interaction with platelet GP1b-IX-V receptors | Phase 2 and 3 trials demonstrated reduced time to platelet normalization, lower exacerbation rates, and reduction in mean PEX procedures and hospital length of stay compared with placebo |
| NAC | Cleaves VWF multimers by reducing disulfide linkages | Reduction in VWF multimer size and VWF-dependent platelet aggregation in preclinical models |
| Case reports of successful use in refractory TTP | ||
| Bortezomib | Reduction in anti-ADAMSTS13 antibody production through apoptotic destruction of plasma cells | Case reports and series of successful use in patients refractory to multiple lines of therapy |
| Recombinant ADAMTS13 | Neutralization of anti-ADAMSTS13 antibodies and restoration of ADAMSTS13 activity | Phase 1 trial demonstrated safety in congenital TTP; phase 3 trial planned |
| Anfibatide | Reduces platelet–VWF interaction through antagonism of the platelet GP1b receptor | Inhibited platelet–VWF aggregate formation in in vitro studies and resolved thrombocytopenia in a mouse model of TTP |
Caplacizumab
Caplacizumab is a bivalent humanized nanobody (a single-variable-domain immunoglobulin) that attaches to the A1 domain of VWF and effectively inhibits its interaction with platelet GP1b-IX-V, reducing platelet aggregation and microvascular thrombosis.8,9 The international phase 2 TITAN study randomized 75 patients with acquired TTP to caplacizumab or placebo in combination with daily PEX and immunosuppression and reported a significantly shortened time to platelet normalization (3.0 days vs 4.9 days) and a lower rate of exacerbations (8% vs 28%) in the caplacizumab group.8 The HERCULES trial (reported at the 2017 meeting of the American Society of Hematology), a randomized double-blind phase 3 study, evaluated the efficacy and safety of caplacizumab in 145 patients with TTP.9 In addition to daily PEX and immunosuppression, patients received a 10-mg IV loading dose of caplacizumab or placebo after PEX initiation, followed by once-daily subcutaneous administration of 10 mg of the study drug or placebo for at least 30 days after PEX discontinuation. Caplacizumab was associated with significantly quicker time to platelet count normalization (platelet normalization rate ratio 1.55, P < .01), and at 5 days, 94% of patients in the caplacizumab group had normalized platelet count compared with 77% in the control group.9 There was a 38% reduction in mean number of PEX procedures (5.8 vs 9.4 days) and a significant shortening in length of intensive care (by 61%) and overall length of stay (by 31%).9 The composite secondary end point of acquired TTP-related death, acquired TTP recurrence, or major thromboembolic event was also significantly reduced (12.7% vs 49.3%, P < .001) in patients receiving caplacizumab.9 The safety profile of caplacizumab was favorable; bleeding events that might be expected based on its mechanism of action were the most common adverse events, as was seen in the TITAN study.8,9 Most events were mild to moderate in severity, and epistaxis was the most common.
The outcomes of the TITAN and HERCULES trial are encouraging, in terms of both faster platelet recovery and reduced exacerbation rate; however, caplacizumab was continued for 30 days beyond the last PEX (the window for exacerbation), and several patients in the caplacizumab arm relapsed in the month after discontinuing caplacizumab. For example, recurrence between 30 days and 12 months occurred in 31% of the caplacizumab group and only 8% of controls in the TITAN study.8 In HERCULES, 9% relapsed within the month after stopping the caplacizumab.9 This highlights the point that caplacizumab has a protective effect in the acute phase of the disease but does not modify the underlying immune pathophysiology of acquired TTP and will still need to be used in combination with immunosuppression. Future studies are needed to identify risk factors for relapse (such as ADAMTS13 activity and anti-ADAMTS13 antibody) on stopping caplacizumab and evaluate whether these can be used to guide duration of therapy. The cost-effectiveness of caplacizumab as a first-line option for all patients, especially for those who also receive upfront rituximab, needs evaluation. Nevertheless, caplacizumab is likely to become the first drug approved for TTP and is an exciting addition to the TTP armamentarium. We are particularly optimistic regarding its role in refractory cases to minimize ischemic injury in patients with evidence of cardiac or neurologic events and as initial treatment along with plasma infusions for patients presenting to hospitals without PEX services before transfer to a PEX-capable facility.
NAC
The mucolytic N-acetylcysteine (NAC) has been explored as an adjunct to standard therapies in TTP due to the structural similarity of disulfide linkages in VWF and polymeric mucins. Pre-clinical studies showed that NAC reduces large VWF multimers and inhibits VWF-dependent platelet aggregation.36 Several case reports suggested that NAC is an effective adjunct in refractory cases of TTP, while others did not report a benefit36,37 ; however, concurrent administration of other agents confounds the interpretation of these results. Caution is warranted in patients with active bleeding, as NAC may increase bleeding through its effect on VWF–platelet interactions or by reducing activity of vitamin K–dependent clotting factors. Further evaluation of the efficacy and safety of NAC as an adjunct to standard therapy in TTP is in progress (NCT01808521).
Plasma cell–directed therapy (bortezomib)
Bortezomib, a proteasome inhibitor, is a widely used therapy for multiple myeloma. It targets plasma cells rather than B cells and has emerged as a promising therapy for refractory TTP. To date, 12 cases (3 relapsed, 9 refractory) treated with bortezomib have been reported.38,39 Although 11 of 12 had clinical responses after bortezomib therapy, all had received prior PEX, corticosteroids, rituximab, and often additional immunosuppressants, which makes it difficult to ascertain the contribution of bortezomib. The efficacy of bortezomib in multirefractory TTP is attributed to the fact that it targets plasma cells that can produce anti-ADAMTS13 autoantibodies even after CD20-positive B cells have been effectively depleted.39 Prospective studies are needed to establish the efficacy and safety of bortezomib in TTP.
rADAMTS13
Recombinant ADAMTS13 (rADAMTS13; SHP655/BAX930) was developed to overcome the adverse effects associated with fresh frozen plasma administration, including allergic reactions, viral infections, and volume overload. In a phase 1 trial of 15 patients with congenital TTP, rADAMTS13 administration was associated with a dose-dependent increase in ADAMTS13 level, a reduction in ultra-large VWF multimers, and minimal adverse effects.40 The major application of rADMATS13 is expected to be in congenital TTP. An open-label phase 3 trial investigating rADAMTS13 vs standard of care in congenital TTP has been initiated (NCT03393975), and the drug has been granted fast-track designation by the US Food and Drug Administration. The role of rADAMTS13 in acquired TTP is more complex. In vitro studies using patient plasma suggest that higher concentrations of rADAMTS13 may be able to overcome the inhibitor and increase ADAMTS13 activity in acquired TTP.41 It has also been hypothesized that rADAMTS13 variants with altered antigenic domains and enhanced enzymatic activity may be an effective therapeutic strategy for acquired TTP.42 Additional studies conducted in human subjects are needed to evaluate the efficacy of rADAMTS13 as a replacement for PEX or as an adjunct to minimize the volume and duration of plasma therapy.
Other novel agents
Anfibatide is a potent platelet GP1b receptor antagonist derived from snake venom, which inhibits platelet aggregation by inhibiting its interaction with VWF. In a microfluidic shear model, anfibatide inhibited platelet adhesion, aggregation, and thrombosis formation under static and shear conditions and resulted in resolution of spontaneous thrombocytopenia in a Shiga toxin–induced murine TTP model.43 However, the safety and efficacy of anfibatide for TTP treatment in human subjects requires further study in prospective clinical trials. Emerging data suggest that TTP may be associated with complement activation, perhaps through ultra-large VWF–mediated effects,44 and the successful use of the terminal complement inhibitor, eculizumab, in TTP has been reported.45
Conclusion
Outcomes of TTP improved dramatically after the introduction of PEX, but there is still a 10% to 15% risk of mortality with each episode. Novel targeted therapies have been developed based on TTP pathogenesis and will potentially improve outcomes of acquired TTP even further. Recent data support that rituximab is beneficial in acute TTP, both upfront and for refractory cases, and is fast gaining traction as prophylactic therapy to prevent relapses in patients that have persistent or recurrent ADAMTS13 deficiency in remission. Caplacizumab has completed clinical trials and is expected to be approved soon. It is interesting to speculate that by reducing dependence of PEX, caplacizumab (perhaps in combination with rADAMTS13 and immunosuppression) could usher in a new paradigm of treating TTP with minimal, if any, PEX. Other novel agents are in development, but prospective studies are needed to establish efficacy and define their optimal use in TTP.
Correspondence: Shruti Chaturvedi, Division of Hematology, Johns Hopkins University School of Medicine, 720 Rutland Ave, Ross Research Building Rm 1025, Baltimore, MD 21205; e-mail: schatur3@jhmi.edu.
