
Abstract
Treatment outcomes for patients with peripheral T-cell lymphomas (PTCLs) and advanced-stage cutaneous T-cell lymphomas (CTCLs) remain poor. The past few years have witnessed an explosion in our understanding of the genetics of these diverse malignancies. Many subtypes harbor highly recurrent mutations, including single-nucleotide variants, insertions/deletions, and chromosomal rearrangements, that affect T-cell receptor signaling, costimulatory molecules, JAK/STAT and phosphatidylinositol 3-kinase pathways, transcription factors, and epigenetic modifiers. An important subset of these mutations is included within commercially available, multigene panels and, in rare circumstances, indicate therapeutic targets. However, current preclinical and clinical evidence suggests that only a minority of mutations identified in TCLs indicate biologic dependence. With a few exceptions that we highlight, mutations identified in TCLs should not be routinely used to select targeted therapies outside of a clinical trial. Participation in trials and publication of both positive and negative results remain the most important mechanisms for improving patient outcomes.
Learning Objectives
Gain understanding of the complexity of genetic lesions identified across the many subtypes of CTCL and PTCL
Learn about approaches to target these lesions and the need for preclinical and/or human trial data prior to noninvestigational use of new therapies
Introduction
T-cell lymphomas (TCLs) comprise ∼10% of all non-Hodgkin lymphomas in western countries. Two-thirds of these lymphomas are peripheral TCLs (PTCLs) and the remainder are cutaneous TCLs (CTCLs).1 Many patients with CTCL have an indolent course that can span decades, although patients with advanced-stage disease have a poor prognosis. In contrast, nearly all PTCLs are aggressive lymphomas. The World Health Organization currently divides TCLs into 29 different subtypes based on histologic and immunophenotypic features that include both αβ and γδ TCLs.2 Emblematic of our poor understanding of the biology of these diseases, the most common subtype of PTCL is “not otherwise specified (NOS).”
Treatment of PTCLs to date has been largely derivative or empiric. Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP)-based regimens, which were adopted based upon their activity in B-cell lymphomas, remain the standard first-line therapy. Except for anaplastic large cell lymphomas (ALCLs) that harbor rearrangements of anaplastic lymphoma kinase (ALK; ALK+ ALCL), over 75% of PTCLs will be refractory to frontline treatment or relapse within 2 years of initial therapy. The most common subtypes of PTCL, including PTCL-NOS, angioimmunoblastic TCL (AITL), ALCL that lacks ALK rearrangements (ALK− ALCL), and natural killer/TCL, as well as advanced-stage CTCL, all have 5-year overall survival rates under 30%. There are very few long-term survivors of rare subtypes of TCL such as hepatosplenic TCL and monomorphic epitheliotropic intestinal TCL (MEITL).
Based on the limited efficacy of current treatments, the National Cancer Comprehensive Network guidelines list participation in a clinical trial as the top choice for all patients with PTCL (other than ALK+ ALCL) in both the firstline and relapsed/refractory settings. Most US Food and Drug Administration (FDA)-approved agents for relapsed/refractory PTCL, such as pralatrexate and the histone deacetylase inhibitors romidepsin and belinostat, have no biomarkers predictive of response. Furthermore, the mechanisms that mediate intrinsic and acquired resistance are both poorly understood and difficult to model. As such, combination studies have mostly used empiric regimens of multiple agents with limited activity at maximum tolerated doses.
Recent advances in the biologic understanding of TCLs
The rational translation of molecular discoveries into informed therapeutic strategies has been extraordinarily successful for B-cell lymphomas. Multiple small molecules targeting B-cell receptor signaling (eg, ibrutinib, idelalisib) and apoptotic vulnerabilities (eg, venetoclax), antibodies targeting immune checkpoints (eg, pembrolizumab, nivolumab) or surface proteins (eg, rituximab), and chimeric antigen receptor T cells targeting CD19 have all improved outcomes for patients with B-cell lymphomas.
There are 2 prominent success stories targeting TCL biology that suggest the same kind of progress is possible for these diseases. The first is brentuximab vedotin, an antibody-drug conjugate that targets CD30 and is highly active in ALCL,3 cutaneous CD30+ CTCL,4 and possibly other TCLs.5 The second is small-molecule inhibitors of ALK for the treatment of ALK+ ALCL.6 Yet, multiple roadblocks have prevented successful discovery and translation for most TCLs: (1) the low incidence of each subtype, which limits sample availability and clinical trial enrollment; (2) heterogeneity across subtypes, which further complicates biologic interrogation; and (3) a lack of faithful model systems for in vitro and in vivo studies.7 Through concerted effort to overcome these roadblocks, a sea change has occurred in TCL research.
Multiple groups have now reported genetic and transcriptional landscapes for individual TCL subsets.8-20 Outstanding reviews of these landscapes were recently published and the reader is referred to them for discussions of individual mutations and their molecular epidemiology.21,22 Instead, we will focus on the practical implications of genetic abnormalities for diagnosis, prognosis, and therapeutic selection. For the sake of brevity and focus, we will primarily discuss PTCLs.
Validating candidate targets in model systems
Unsurprisingly, genetic alterations in TCLs commonly affect pathways that mediate T-cell activation, proliferation, and subtype-specific identity, most notably the pathways downstream of the T-cell receptor (TCR), costimulatory proteins, cytokine receptor signaling through JAK/STAT and phosphatidylinositol 3-kinase pathways, transcription factors, and epigenetic modifiers (Figure 1). Among these, activated kinases are attractive targets from the perspective of chemical biologists, as evidenced by the large number of kinase inhibitors used in other cancers.23
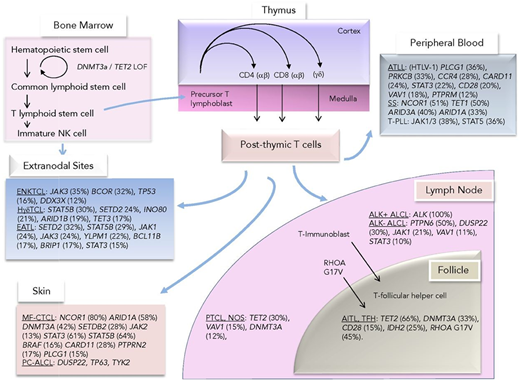
Mutations observed across TCLs divided by their presumed cell of origin or closest immunophenotypic counterpart. Reprinted from Van Arnam et al.21 ATLL, adult T-cell leukemia/lymphoma; EATL, enteropathy-associated TCL; ENKTCL, extranodal natural killer/TCL; HγδTCL, hepatosplenic γ-δ TCL; MF-CTCL, mycosis fungoides CTCL; PC-ALCL, primary cutaneous ALCL; TFH, T-follicular helper cell; T-PLL, T-cell prolymphocytic leukemia.
Mutations observed across TCLs divided by their presumed cell of origin or closest immunophenotypic counterpart. Reprinted from Van Arnam et al.21 ATLL, adult T-cell leukemia/lymphoma; EATL, enteropathy-associated TCL; ENKTCL, extranodal natural killer/TCL; HγδTCL, hepatosplenic γ-δ TCL; MF-CTCL, mycosis fungoides CTCL; PC-ALCL, primary cutaneous ALCL; TFH, T-follicular helper cell; T-PLL, T-cell prolymphocytic leukemia.
Dysregulation of kinase activity occurs in essentially all TCL subtypes through overexpression, mutation, amplification, or rearrangement (Figure 1).24 For example, there are multiple SRC family kinases such as FYN and the Tec family kinase interleukin-2 (IL-2)–inducible T-cell kinase (ITK) downstream of the TCR (Figure 2). Somatic-activating mutations of FYN have been identified in PTCLs and CTCLs.10,20,25 The kinase SYK is overexpressed in a large fraction of PTCLs relative to normal T cells.26 Fusions of ITK to the kinase domain of SYK occur in a subset of PTCLs and result in a constitutively active kinase.27 Mutations affecting the MAPK pathway, including activating mutations in KRAS and MEK (MAP2K1), occur in a variety of TCLs.21 Nearly all TCLs have biochemical evidence of constitutive JAK/STAT signaling. A subset have bona fide activating mutations or chromosomal rearrangements of JAK or STAT genes.11,16,18,19,28
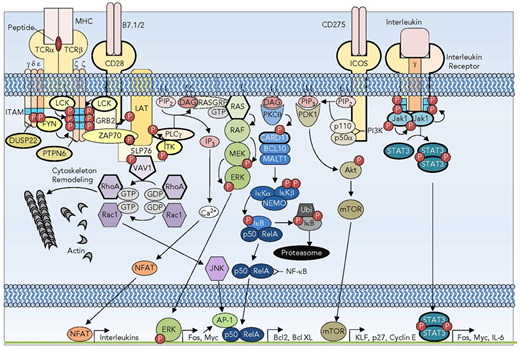
Signaling pathways subverted in T-cell oncogenesis. A subset of the TCR, costimulatory, and interleukin receptor pathways showing the redundancy and complexity of signaling. Recurrently mutated receptors and signaling pathway components are depicted with bold outlines. DAG, diacylglycerol; GTP, guanine nucleotide triphosphate; IL-6, interleukin-6; IP3, inositol 3-phosphate; MHC, major histocompatibility complex; mTOR, mammalian target of rapamycin; P, phosphorylation; PTPN, protein tyrosine phosphatase nonreceptor. Reprinted from Van Arnam et al.21
Signaling pathways subverted in T-cell oncogenesis. A subset of the TCR, costimulatory, and interleukin receptor pathways showing the redundancy and complexity of signaling. Recurrently mutated receptors and signaling pathway components are depicted with bold outlines. DAG, diacylglycerol; GTP, guanine nucleotide triphosphate; IL-6, interleukin-6; IP3, inositol 3-phosphate; MHC, major histocompatibility complex; mTOR, mammalian target of rapamycin; P, phosphorylation; PTPN, protein tyrosine phosphatase nonreceptor. Reprinted from Van Arnam et al.21
Many somatic alterations in nonkinase genes can also activate kinase signaling in TCLs. These include loss of promoter methylation and loss of expression among phosphatases (eg, protein tyrosine phosphatase, non-receptor type 6/Src homology region 2 domain-containing phosphatase 1 and protein tyrosine phosphatase, receptor type K) that directly antagonize the same kinases that are recurrently mutated in TCLs (Figure 2).1,2 Mutations in the chemokine receptor CCR4 occur in 25% of adult T-cell leukemia/lymphomas (ATLLs) and some other TCLs.29 These mutations impair receptor internalization and increase PI3K pathway signaling. Signaling through PI3K/mammalian target of rapamycin is also upregulated by recurrent alterations in Rho GTPases and guanine exchange factors (GEFs) in TCLs.3-5 Most notably, up to 60% of angioimmunoblastic TCLs harbor a recurrent RhoA G17V mutation.10,17 Other RhoA mutations are observed in CTCL, ATLL, and PTCL-NOS. Mutations involving the guanine exchange factor VAV1 occur in several subtypes of TCL and include single-nucleotide variants (SNVs), in-frame deletions, and fusions (Figure 1).30
With all of these therapeutic targets, why are there no better treatments for TCL? The obvious answer is that the vast majority of these targets, despite mutation, overexpression, amplification, and even biochemical evidence of pathway activation, are not true vulnerabilities. Previous efforts to advance therapeutic strategies based solely on expression or mutation have largely failed. For example, platelet-derived growth factor receptor (PDGFR) was reported to be highly expressed in PTCL-NOS, to drive an autocrine regulatory pathway in PTCL cell lines, and even to confer a targetable vulnerability in a mouse model of ALK+ lymphoma.6-8 Thus, we performed a trial of imatinib, which is active in vitro against PDGFR and has extraordinary activity against FIP1L1-PDGFRα–rearranged cells,31 in patients with TCL. The response rate among 12 patients was 0% and median progression-free survival was 21 days.32
There are, of course, many reasons why a clinical trial can fail. Figure 2 captures only a few of the diverse and complicated signaling pathways that drive proliferation and survival within T cells. Although there are many druggable targets within these pathways, the remarkable redundancy is likely to limit absolute dependence on nearly all single alterations. In addition, many mutations or other genetic events are present within subclones, so effective targeting is unlikely to elicit deep responses.
How then, do we clarify which alterations represent optimal therapeutic targets? There are no definitive answers to that question. One possibility is that certain types of alterations are more important than others. For example, both SNVs and fusions involving JAK2 occur in B-cell leukemias. The substitutions (eg, JAK2 R683G) appear to confer little if any vulnerability to the JAK2 inhibitor ruxolitinib.33,34 In contrast, ruxolitinib can induce complete responses in patients with leukemia harboring JAK2 fusions, akin to those observed in patients with fusions involving ABL or PDGFR who are treated with imatinib.35
To extend these observations to TCLs, we and others are assessing the functional relevance of individual alterations using patient-derived xenograft (PDX) and transgenic models.10,3,4,28,36-38 Several transgenic models of TCL have been published, including lymphomas induced by ITK-SYK, Roquin deletion, RhoA G17V, loss of programmed death 1 (PD1) or Tet methylcytosine dioxygenase 2 (TET2), overexpression of IL-15, and other recurrent abnormalities observed in patients.3-5,39-41 Over 50 PDX models spanning a large number of TCL subtypes have been serially propagated in immunocompromised mice and many are available to the research community through our Public Repository of Xenografts (http://www.proxe.org).36,37 The Inghirami laboratory used PDX models to demonstrate that, like B-cell leukemias, SNVs in JAKs and STATs in human ALK− ALCLs do not confer absolute dependence on JAK/STAT signaling.28 In contrast, JAK2 fusions within a CTCL cell line and a T-cell prolymphocytic leukemia (T-PLL) PDX resulted in extraordinary sensitivity to ruxolitinib (Figure 3).36
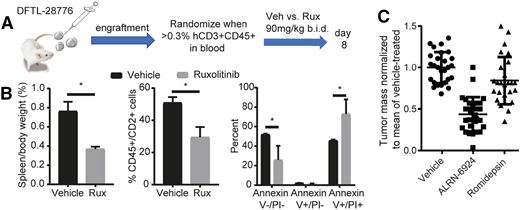
Examples of genetically informed therapies in PTCL. (A) Protocol for testing the JAK2 inhibitor ruxolitinib (Rux) in a T-PLL PDX with a BCR-JAK2 fusion. (B) Reduced spleen size and spleen disease burden, and increased apoptosis (Annexin V+) in residual T-PLL cells at day 8 of treatment. *P < .05 by 2-sided Student t test with Welch correction. (C) Pooled data showing consistent activity of the MDM2/MDMX inhibitor ALRN-6924 across 5 different p53–wild-type PTCL PDXs (5 mice per arm per PDX) after 8-day treatment. Each point indicates a mouse. Tumor was subcutaneous xenograft in 2 models or splenic in 3 disseminated, orthotopic models. Data reproduced from Ng et al with permission.36
Examples of genetically informed therapies in PTCL. (A) Protocol for testing the JAK2 inhibitor ruxolitinib (Rux) in a T-PLL PDX with a BCR-JAK2 fusion. (B) Reduced spleen size and spleen disease burden, and increased apoptosis (Annexin V+) in residual T-PLL cells at day 8 of treatment. *P < .05 by 2-sided Student t test with Welch correction. (C) Pooled data showing consistent activity of the MDM2/MDMX inhibitor ALRN-6924 across 5 different p53–wild-type PTCL PDXs (5 mice per arm per PDX) after 8-day treatment. Each point indicates a mouse. Tumor was subcutaneous xenograft in 2 models or splenic in 3 disseminated, orthotopic models. Data reproduced from Ng et al with permission.36
This does not mean that some SNVs or insertions/deletions are not valuable biomarkers for targeted therapy, but instead that several fusions and other structural variants have simply risen to the top of the current list of important targets. Clinical therapeutics that have been validated within patients and/or models harboring specific fusions, include: (1) SYK, ALK, and JAK inhibitors for fusions involving these kinases6,19,40 ; (2) inhibitory antibodies against CTLA4 for CTLA4-CD28 fusions42 ; and (3) inhibitors of PD1 interactions for structural variants involving the 3′ untranslated regions (UTRs) of PD ligand 1 (PD-L1) or PD-L2.12,14
Identifying additional genetic lesions that predict therapeutic response should be a priority, as should reporting of both successful and unsuccessful efforts to target them. The latter is particularly important because publication bias toward responders leads to inaccurate perceptions of drug efficacy, which drives the inappropriate use of ineffective and potentially toxic therapies. At the same time, modern trial designs are needed to highlight the most promising new agents and combinations based on early signals that guide “go/no-go” decision-making.
Even in PTCLs that lack a targetable genetic lesion such as a kinase fusion, there may still be genetic information that can help guide selection for clinical trials. For example, our laboratory demonstrated that the MDM2/MDMX inhibitor ALRN-6924 is highly active in vivo across multiple p53–wild-type PDX models of TCL (Figure 3).36 As expected for an inhibitor of MDM2/MDMX, cells that harbored p53 mutations were completely resistant to ALRN-6924. Thus, p53 mutation status is an important biomarker for selecting patients for the trial (NCT02264613).
The biology defined within preclinical models can be an important determinant of clinical trial selection and design. The complexity of TCLs highlights the need to interrogate a broad array of cell lines and in vivo models derived at various phases of treatment. Clinical testing of a novel therapy or combination of therapies without careful preclinical testing may not only be ineffective but may also be dangerous, as evidenced by the explosive growth of ATLL reported in some patients treated with PD1 inhibitors.43 The primary shortcoming of our preclinical models is the failure to capture microenvironmental and immune interactions that exist within humans. Thus, it remains axiomatic that drugs may have very different effects in preclinical models and in patients. Overreliance on preclinical models does risk both unwarranted development of inactive compounds and premature abandonment of potentially efficacious therapies. Newer approaches that use humanized mice may offer advantages but at present, there is no substitute for samples collected directly from treated patients.
Genomically targeted therapy for patients with TCL in 2018
Despite the genetic insights noted previously, the selection of therapy for most patients with relapsed/refractory TCL remains empiric. Genetic lesions that should seemingly predict therapeutic responses typically do not. For example, one might surmise that mutations of TET2, which affect DNA methylation and are found in almost 80% of AITLs and other follicular helper TCLs,10,13,44 would predict the likelihood of response to treatment with the demethylating agent azacytidine. In fact, the response rate to azacytidine in these lymphomas may be very high45 but preliminary reports indicate no association between response and TET2 mutation status. Similarly, we report at this American Society of Hematology (ASH) meeting that the response rate to ruxolitinib in an ongoing phase 2 trial of patients with TCL does not seem to differ between patients with or without JAK/STAT mutations.
In many circumstances, it is overly simplistic to infer a direct relationship between an identifiable mutation and a therapeutic response to an inhibitor of the pathway in question. In the example of JAK/STAT mutations, pathway activation may occur in the absence of identifiable mutations (eg, through cytokine interactions within the microenvironment) and lead to similar biology within TCLs that harbor or lack mutations in the pathway. In the example of TET2 and demethylating agents, the differentiation state of the tumor (eg, a T-follicular helper cell–like cell for all AITLs) may confer addiction to DNA methylases that can be exploited independently of TET2 mutation status. Therefore, identifying the context and the biological consequences of a mutation are equally if not more important than identifying the mutation itself when determining the implications of a mutation for therapeutic targeting.
Participation in a well-designed clinical trial remains the most attractive option for most patients with either untreated or previously treated TCL. At this ASH meeting, several groups will report partial and complete response rates >50% among patients with relapsed/refractory TCL receiving investigational agents or combinations. When a trial option is not available, the poor prognosis of patients with relapsed/refractory PTCL and late-stage CTCL makes it reasonable to consider off-label or compassionate use of available agents, but only if there is compelling preclinical and/or patient evidence suggesting benefit. This was evident when crizotinib first became available for the treatment of ALK-rearranged lung cancer, and patients with relapsed ALK+ ALCL received the drug. It remains evident for some of the kinase fusions, including those involving JAK1, JAK2, and CTLA4. Unfortunately, such compelling evidence is lacking for almost all genetic alterations found in TCLs.
There are now commercially available, multigene platforms (eg, Archer DX FusionPlex Lymphoma) that can identify fusions, mutations, and gene expression from formalin-fixed, paraffin-embedded specimens. The same assays can also identify genetic alterations with prognostic relevance, including rearrangements of DUSP22 and TP63. Rearrangements involving the DUSP22 locus are present in ∼30% of ALK− ALCLs and confer a favorable prognosis after CHOP-based chemotherapy.46 In contrast, rearrangements involving TP63 occur in a smaller percentage of TCLs (and B-cell lymphomas)47 and confer a dismal prognosis. Thus, it is a reasonable option to submit TCL biopsies for multigene testing with a particular focus on gene fusions. As more information is gained about the relationships between mutations and treatment response, this may become a standard of care.
In conclusion, we have made tremendous progress in our understanding of TCL genomics while establishing preclinical models that capture some of that biology. There are now a multitude of new therapeutics making their way through the drug development pipeline. The next challenge is to establish biologically informed predictors of response that match individual patients with the most effective therapy. To do this will require that we discard simplistic models of disease biology and abandon the reflexive tendency to equate shared histology or shared mutations as the sole predictors of susceptibility to targeted therapy. As always, equipoise remains paramount.
Acknowledgments
The authors thank Steven Horwitz for careful review of the manuscript. The authors also thank Kojo Elenitoba-Johnson and Megan Lim for allowing use of their outstanding publication figures.
Correspondence
David M. Weinstock, Dana-Farber Cancer Institute, 450 Brookline Ave, Dana 510B, Boston, MA 02215; e-mail: davidm_weinstock@dfci.harvard.edu.
