
Abstract
Although almost 90% of children with acute lymphoblastic leukemia (ALL) and ∼60% of children with acute myeloid leukemia are cured with frontline therapy, relapse and chemotherapy resistance are significant challenges that contribute to morbidity and mortality. Even with long-term survival, the acute and chronic burdens of therapy are major issues for patients and families. Long-term side effects occur, including cardiac, endocrinologic, neurcognitive, orthopedic, and psychosocial problems, and healthy survivorship is frequently compromised. With goals of minimizing relapse and/or decreasing traditional chemotherapy-associated toxicities, exploration of immunotherapeutic strategies has moved to the forefront in pediatric cancer. New immunotherapy approaches provide a major paradigm shift in oncology overall, often curing previously incurable patients. The past several years have yielded successful uses across a variety of malignancies, and enthusiasm continues to rise for applying these therapies more broadly. Herein we discuss current approaches incorporating the bispecific T-cell engager blinatumomab, the antibody-drug conjugate inotuzumab ozogamicin (InO), and CD19-directed chimeric antigen receptor T cells in children with relapsed/refractory B-cell ALL and discuss the potential for using these immunotherapies in the treatment of newly diagnosed children.
Learning Objectives
Describe results of studies with blinatumomab, InO, and CD19-directed CAR-T cells in pediatric refractory and relapsed ALL
Describe strategies for moving blinatumomab, InO, and CD19-directed CAR-T cells to the front line in pediatric ALL
Clinical case
A 14-year-old girl is diagnosed with high-risk B-cell acute lymphoblastic leukemia (ALL; B-ALL) after 2 months of progressive generalized symptoms. While awaiting cytogenetic and molecular results, 4-drug induction chemotherapy is started. Induction therapy is complicated by hyperglycemia necessitating insulin, an anaphylactic reaction to polyethylene glycol (PEG)-aspargase, and necrotizing pancreatitis requiring surgical intervention. Cytogenetics from diagnostic marrows show near haploidy with 26 chromosomes. End-induction minimal residual disease (MRD) is 1.2%. The patient and family are told that these particular findings portend an extremely poor prognosis with conventional chemotherapy alone. After extensive review of several treatment and supportive/palliative care options, the patient receives blinatumomab achieving MRD− status after 28 days of therapy, an additional course of blinatumomab during stem cell transplant (SCT) workup, and, subsequently, an 8 of 8 HLA-matched hematopoietic SCT (HSCT) with curative intent.
Introduction
The overall outcomes for children with ALL have improved dramatically over the past 40 years, with each successive generation of clinical trials showing improvements and refinements of direct and supportive care therapies.1 The vast majority of this progress is not due to the introduction of new agents into therapy regimens but improving survival through therapy intensification. A consequence of intensification is that substantial acute and chronic toxicities continue to accumulate in survivors and this burden of therapy extends to families and the larger community, potentially for many decades if a survivor of childhood cancer lives a full life expectancy. Advances in drug development and introduction of new agents have been historically slower to reach pediatric patients but recently there has been an acceleration in availability and regulatory approval for novel agents for childhood leukemia treatment.
Three of these promising approaches are the CD3/CD19 bispecific T-cell engager blinatumomab, the CD22-directed antibody drug conjugate inotuzumab ozogamicin (InO), and CD19 chimeric antigen receptor T-cell (CAR-T) therapy; these are the focused compounds of this review although many more agents are under active investigation. Both blinatumomab and CAR-T cells discussed here target the CD19 antigen, which is highly expressed on B cells throughout development, in >90% of B-cell lineage cancers and virtually all childhood precursor B-ALL. Both blinatumomab and CAR-T cells essentially activate the endogenous immune response by engaging a patient’s own T cells to seek and destroy CD19+ leukemic blasts. In this process, T cells are activated and may unleash not only an aggressive immune response against the leukemia, but also with what is essentially an on-target immune activation that can lead to substantial toxicity with cytokine release syndrome, an inflammatory response to immune activation and leukemic cell destruction, and less well-understood neurologic toxicities. Both of these side effects have been severe enough in some patients to lead to formal Risk Mitigation and Evaluation Strategies (REMS) and boxed warnings on drug labels; however, both can also usually be managed with exquisite supportive care and appropriate intervention based on severity of symptoms and side effects. Additional toxicities of transient but possibly prolonged cytopenias, infection, and hypogammaglobulinemia are frequent although also they represent on-target effects. In fact, hypogammaglobulinemia is often considered a surrogate marker of CAR-T cell persistence and can be readily managed with supportive immunoglobulin replacement.
The antibody-drug conjugate InO is also associated with its own unique toxicity profile, primarily the observation of hepatic toxicity including veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS). This is thought to be at least in part due to the calicheamicin component of this molecule. Other toxicities including transaminase elevation and cytopenias, occasionally prolonged, and are also observed.
Several groups of children with ALL who do not have as substantial an improvement in outcome include but are not limited to those with high-risk/very high-risk genetic features, patients with persistent residual disease after induction/consolidation, and children with Down syndrome (DS). These patients, although having improved survival over the past several decades, still do not experience the excellent outcomes seen in children with molecularly negative disease after remission induction and the most favorable biological features. As such, newer therapies have been explored in these higher-risk populations and several have shown particular promise including unparalleled single-agent activity for patients with relapsed and refractory disease. Nonoverlapping toxicity profiles compared with more conventional cytotoxic agents in use suggest that introducing these newer approaches could better the chances for those at greatest risk for poor outcomes and may reduce at least some morbidity and mortality for what has become an otherwise highly curable disease for many other patients.
CD19-directed antibody therapy in the front line
Blinatumomab is a bispecific T-cell–engaging antibody that brings CD19+ leukemic blasts in proximity to a patient’s own CD3+ T cells, inducing cell lysis and T-cell activation, which in turn leads to CD19+ cell death and the potential for T-cell expansion. It has a very short half-life and is given by continuous infusion for 28 days per cycle. Based on substantial activity in patients with relapsed and refractory B-lineage ALL, the US Food and Drug Administration (FDA) granted accelerated approval of blinatumomab in Philadelphia chromosome–negative (Ph−) relapsed or refractory B-ALL in December 2014.2 This was extended to include full approval for Ph+ and Ph− pediatric patients with relapsed/refractory B-ALL in July 2016. Subsequent approvals in the European Union, the United Kingdom, Japan, Canada, and other countries followed. As of March 2018, blinatumomab received accelerated FDA approval for use in both adults and children in first or second remission but with detectable MRD at or above 0.1%.3 In the United States, this was the first approval in 12 years for a drug in childhood ALL and marked the first regulatory approval for an immune-based therapy for pediatric ALL.
Blinatumomab data in adults
The German Study Group for Adult ALL published results from 2 phase 2 trials of open-label, single-arm blinatumomab for adults with relapsed or refractory ALL. In the first study, 36 patients received blinatumomab for 4 weeks per cycle, with 2 weeks off in between cycles.4 The trial prescribed 2 cycles of remission induction with up to 3 consolidation cycles of blinatumomab allowed for responders, or the option of HSCT. Of the 36 patients treated, 25 (69%) achieved complete remission with (CR) or without (CRh) count recovery, compared with cited historical response rates of 31% to 44%. Eighty-eight percent of responders became MRD− as measured by quantitative polymerase chain reaction of immunoglobulin rearrangements. Thirteen of the 25 responders proceeded to HSCT. Median relapse-free survival (RFS) in this study was 7.6 months; median overall survival (OS) was 9.8 months, a substantial improvement compared with historical controls. Two of 13 patients who received post-blinatumomab HSCT relapsed; 8 of 12 non-HSCT patients relapsed.
A second multi-institutional phase 2 single-arm study enrolled 189 patients.5 CR/CRh rate in this cohort of patients was 43%; 82% of these patients achieved MRD negativity. Forty percent of responders went on to receive allogeneic HSCT. Median OS was 6.1 months. Gökbuget et al compared these outcome data to historical controls from the United States and Europe (694 patients with CR data and 1112 with OS data); weighted analysis revealed a comparator CR rate of 24% and median OS of 3.3 months for this large control cohort compared with the findings in the phase 2 blinatumomab trial.6 The odds ratio of CR was 2.68 (1.67-4.31) in favor of blinatumomab, and the OS hazard ratio was 0.536 (0.394-0.730), again associating treatment with blinatumomab with prolonged survival compared with standard cytotoxic chemotherapies.
The culmination of the adult ALL data to date was publication of results from the TOWER study, a multi-institutional multinational phase 3 trial that randomized heavily pretreated adults with relapsed/refractory ALL 2:1 to blinatumomab or 1 of 4 standard-of-care chemotherapy regimens.7 Four hundred five patients were enrolled, and 376 received at least 1 dose of their assigned therapy (267 received blinatumomab, 109 received chemotherapy). The trial was closed early due to an obvious benefit in the blinatumomab arm. Patients receiving blinatumomab experienced CR rates double those receiving chemotherapy (34% vs 16%; P < .001) and exhibited a significant, twofold improvement in median OS (7.7 months vs 4.0 months; P = .01). Duration of remission and 6-month event-free survival (EFS) were also significantly better in the blinatumomab group (7.3 vs 4.6 months and 31% vs 12%, respectively). The benefit of blinatumomab over standard cytotoxic chemotherapy was most pronounced in those patients for whom trial therapy constituted their second or less salvage therapy and those who had no prior HSCT, suggesting that earlier use of blinatumomab may be more efficacious.
Additional studies have evaluated the use of blinatumomab for adults with high-risk ALL in remission, but with detectable measurable residual disease (MRD+). One phase 2 trial treated MRD+ adult ALL patients with blinatumomab preallogeneic HSCT.8 The trial prescribed 1 cycle of blinatumomab with an option for up to 3 additional cycles for responders; there was also the option to go to HSCT at the treating physician’s discretion. Sixteen of 20 evaluable patients (80%) became MRD−, all after 1 cycle of blinatumomab. Twelve of these 16 responders had never achieved MRD negativity with prior therapies. Eight responders proceeded to HSCT with no relapses reported in the initial study. Long-term follow-up of this cohort was recently reported, with a median follow-up duration of 50.8 months.9 The 5-year EFS for all patients was 50%, superior in comparison with generally accepted outcomes of ≤25% for adult patients with B-ALL and residual MRD.
New data from the most recently published adult study of blinatumomab in the persistent MRD setting provide support for using multiple cycles.9 Although the majority of the MRD responses below the level of detection were seen after 1 cycle, of the 20% of patients who did not achieve MRD negativity after 1 cycle, 10% achieved MRD negativity with a second cycle of therapy. Perhaps most convincingly, one-third of patients on the trial did not receive HSCT or any additional therapy after receiving 3 or more cycles of blinatumomab; these patients had identical survival to those who underwent HSCT. This suggests that even in patients achieving MRD negativity, additional blinatumomab may eradicate disease below the limits of detection.
Blinatumomab data in pediatrics
The pediatric experience with blinatumomab has also been promising. von Stackelberg et al published the results of MT-205 (Children’s Oncology Group [COG] AALL1121), a phase 1/2 dose-escalation/dose-expansion trial of blinatumomab for relapsed or refractory B-ALL in children <18 years of age.10 Eligible patients had primary refractory disease, ALL in second or greater relapse, or relapsed disease after HSCT. The study enrolled 49 patients in phase 1 and 44 patients in phase 2. The pediatric recommended phase 2 dose (RP2D) was determined to be 5 μg/m2 per day for the first 7 days, then 15 μg/m2 per day thereafter. Seventy patients received blinatumomab at the RP2D, and of these, 27 (39%) achieved CR within 2 cycles, with 52% of responders deemed MRD−. Median RFS was 4.4 months, median OS was 7.5 months, and the 6-month RFS was 42%. CR rates were noted to be higher in younger patients, those without primary refractory disease, and those with lower bone marrow blast burden. When restricted to patients with relapsed disease (ie, not refractory), the CR rate was 48%. Given the unfavorable characteristics of the cohort (>70% of patients had relapsed within 6 months of the previous treatment attempt), this response rate was highly encouraging. Long-term follow-up for this study was recently published.11 Fourteen (20%) of the 70 patients who received blinatumomab at the RP2D were alive at 24 months of follow-up; an additional 8 patients (11.4%) were alive at study closure and last known follow-up. Allogeneic HSCT either before or after blinatumomab was associated with improved survival.
With these data and experiences, blinatumomab is an excellent candidate for pediatric investigational studies in B-lineage ALL because it is the first readily available agent introduced as monotherapy that has had substantial capacity to induce MRD− remissions in B-lineage ALL. Adult and pediatric data have shown that patients with lower leukemia burden (<50% bone marrow blasts) at the time of administration have less toxicity and enhanced disease response, thus lending further support to use in patients in remission.9 In adults achieving hematologic CR but MRD positivity, blinatumomab induced MRD− state in 78%.9 Because the current outcomes for children with standard-risk therapy are excellent with tolerable toxicity, any new agent introduced should enhance efficacy without increasing toxicity.
Preliminary data from randomized trials in both the IntReALL (study IntReALL2010/MT215) and COG groups (study AALL1331; shown in Figure 1A) using blinatumomab in the treatment of children with relapsed B-ALL to date show favorable toxicity compared with those who receive conventional multiagent chemotherapy including lower incidences of fever/neutropenia, mucositis, and fewer infections, especially grades 3-5 (A. von Stackelberg, May 2019 and P. Brown, July 2019, oral communications). Although a direct translation from the setting of relapse/refractory disease to frontline therapy is not guaranteed, the paradigm on testing new agents in the relapsed/refractory setting prior to moving forward is one that has met with success in prior generations of clinical trials for childhood and adult ALL, and has met with regulatory and academic support globally. The observation of less infectious toxicity with blinatumomab in the relapse setting prompts the consideration of whether blinatumomab can improve the outcomes by decreasing treatment-related morbidity and mortality for patients with DS who are known to have high rates of infection throughout conventional chemotherapy regimens.
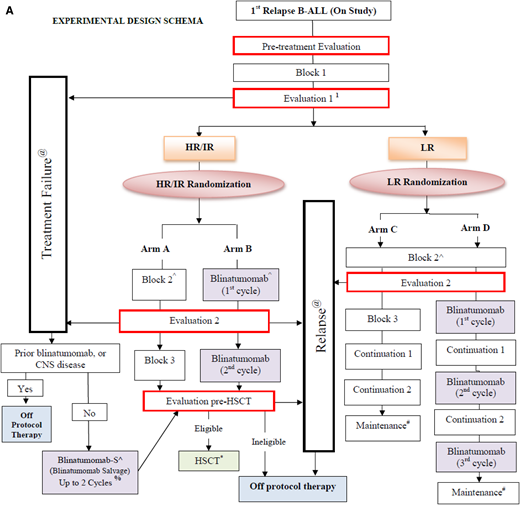
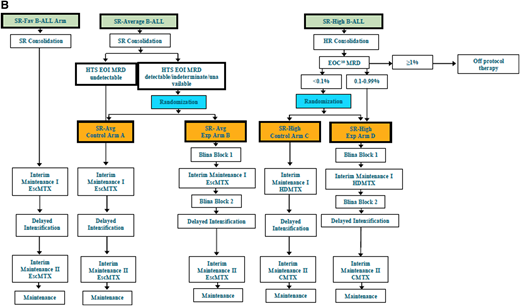
Schema of COG protocols. (A) Schematic diagram of COG study AALL1331 showing the 4 arms of therapy and randomization time points for patients with first relapse of B-ALL to receive blinatumomab. Patients receive a common induction (block 1) and are then risk stratified based on disease response and biologic features. Those with high risk and intermediate risk receive arms A or B. Patients with low-risk disease are randomized to arms C and D. Time points for additional evaluations and consideration of stem cell transplant are depicted as are points where patients would be removed from treatment of nonresponse or relapse. (B) Schematic diagram of COG study AALL1731 showing the 2 control and 2 experimental arms taken from the AALL1331 design with additional biologic testing at defined time points after induction chemotherapy. Patients with SR-Avg and SR-high disease are randomized after consolidation and based on MRD assessments to receive 2 blocks of blinatumomab intercalated into conventional chemotherapy backbone treatment. Blina, blinatumomab; EscMTX, escalating-dose methotrexate; Exp, experimental; SR-Fav B-ALL = standard-risk favorable B-ALL; SR-Avg B-ALL = standard-risk average B-ALL; SR-High B-ALL, standard-risk high B-ALL. Schematic flow diagrams shown with permission of COG AALL1331 Study Chair Patrick Brown and AALL1731 Study Chairs Rachel Rau and Sumit Gupta.
Schema of COG protocols. (A) Schematic diagram of COG study AALL1331 showing the 4 arms of therapy and randomization time points for patients with first relapse of B-ALL to receive blinatumomab. Patients receive a common induction (block 1) and are then risk stratified based on disease response and biologic features. Those with high risk and intermediate risk receive arms A or B. Patients with low-risk disease are randomized to arms C and D. Time points for additional evaluations and consideration of stem cell transplant are depicted as are points where patients would be removed from treatment of nonresponse or relapse. (B) Schematic diagram of COG study AALL1731 showing the 2 control and 2 experimental arms taken from the AALL1331 design with additional biologic testing at defined time points after induction chemotherapy. Patients with SR-Avg and SR-high disease are randomized after consolidation and based on MRD assessments to receive 2 blocks of blinatumomab intercalated into conventional chemotherapy backbone treatment. Blina, blinatumomab; EscMTX, escalating-dose methotrexate; Exp, experimental; SR-Fav B-ALL = standard-risk favorable B-ALL; SR-Avg B-ALL = standard-risk average B-ALL; SR-High B-ALL, standard-risk high B-ALL. Schematic flow diagrams shown with permission of COG AALL1331 Study Chair Patrick Brown and AALL1731 Study Chairs Rachel Rau and Sumit Gupta.
The newest COG frontline protocol for standard-risk B-ALL and B-lymphoblastic lymphoma, AALL1731, randomizes patients to 2 blocks of blinatumomab vs conventional chemotherapy (Figure 1B). Patients with DS-ALL are eligible for focused arms of this study. In one of the primary objectives of the trial, the addition of 2 cycles of blinatumomab to standard therapy will be evaluated to determine whether it can improve disease-free survival (DFS) in patients with standard-risk (SR) B-ALL and selected higher-risk features, and in patients with SR average (SR-Avg) B-ALL who are negative for MRD by flow cytometry but have detectable or indeterminate MRD as measured by high-throughput sequencing at end of induction (Figure 1). On this trial, blinatumomab will be given in 28-day continuous infusion cycles, followed by a 1-week break prior to the initiation of the next cycle of therapy. One of several additional secondary objectives of this trial will be to evaluate the caregiver burden and quality-of-life assessments for patients due to the unique administration demands for continuous blinatumomab infusion delivery on the 28-day schedule.
Additional trials using blinatumomab are beyond the scope of this review, but blinatumomab is increasingly being used in relapse and high-risk settings including as post stem cell transplant consolidative therapy and in several first relapse settings. The International Berlin-Frankfurt-Muenster Consortium (iBFM) proposed IntReALL2020 trial includes blinatumomab following treatment with a CD22-directed agent as consolidation before HSCT for a subset of patients (A. von Stackelberg and F. Locatelli, oral communication, May 2019) and the COG will explore blinatumomab in combination with PD-1 targeted treatment to attempt to overcome CD19− antigen loss (S. Cooper, oral communication, June 2019).
CD22-directed therapies in the front line
Similar to the successes seen with blinatumomab for CD19+ ALL, the CD22 antigen has been successfully targeted for over a decade, including pediatric clinical trials with moxetumomab pasudotox,12 epratuzumab,13,14 and inotuzumab, but not until InO received regulatory approval in August 2017 by the FDA was a CD22-directed agent widely available for use and further testing in children.15 For the purposes of this review, moxetumomab and epratuzumab are not addressed, as there are no active development efforts for these drugs in childhood ALL currently. However, moxetumomab pasudotox was recently approved in the United States for relapsed/refractory hairy cell leukemia, and epratuzumab for second-line use in B-cell chronic lymphocytic leukemia. Results of the IntReALL 2010 study including epratuzumab will be presented at a later date (A. von Stackelberg, oral communication, February 2019).
Adult data with InO
Much in line with the approach using blinatumomab initially in relapse/refractory disease, InO has undergone testing in children after the successful clinical trials in adults. Data supporting FDA approval came largely from the INO-VATE ALL trial,15 a phase 3 trial in which adults with relapsed/refractory B-ALL were randomized to receive single-agent InO on days 1, 8, and 15 in 21- or 28-day cycles vs standard intensive chemotherapy as first or second salvage therapy. Primary end points for the trial were CR with or without count recovery and OS. The first 218 patients (109 per treatment group) of 326 total patients randomized were evaluated in an intent-to-treat analysis for CR. The researchers found significantly higher CR rates in the InO group compared with the standard chemotherapy group (80.7% vs 29.4%). Other favorable outcome measures were also higher in the InO group including MRD− rates (78.4% vs 28.1% for the chemotherapy group) and longer duration of remission (4.6 months vs 3.1 months for the chemotherapy group) in those who achieved CR. Survival analysis of all 326 patients was also performed: progression-free survival (PFS) was 5 months for InO vs 1.8 months for chemotherapy and median OS was 7.7 months for InO vs 6.7 months for chemotherapy (P, .04). The least amount of benefit of InO over standard chemotherapy was seen in Ph+ and t(4;11) patients. This trial also noted a significantly higher incidence of SOS with InO, which has been a consistently observed toxicity in adult patients treated with this drug.
In addition to this large phase 3 trial, there have been several published single-institution trials of InO usage in relapsed/refractory ALL.16-18 MD Anderson published their experience with InO as a single dose16,17 or with weekly dosing17 in a total of 90 patients, 68% of whom were on their second or higher salvage therapy for ALL. InO was given on day 3 of the first 4 cycles. Induction chemotherapy was a mini-hyper-CVD regimen consisting of cyclophosphamide, vincristine, and dexamethasone. Odd-numbered cycles when InO was combined with chemotherapy included cyclophosphamide (150 mg/m 2 IV every 12 hours on days 1-3), oral or IV dexamethasone (20 mg per day on days 1-4 and days 11-14), and vincristine on days 1 and 8. Even-numbered cycles used methotrexate (250 mg/m2 IV on day 1) and cytarabine (0.5 g/m 2 IV every 12 hours on days 2 and 3). Maintenance therapy with 6-mercaptopurine, vincristine, oral methotrexate, and prednisone was given for 3 years, although the primary end point was PFS at 2 years.
Overall response rate for this cohort of patients was 58%, of whom 19% achieved CR and 30% achieved CRh. Of the 50 responding patients, 72% became MRD− with InO; no differences were reported between response rates in single-dose vs weekly dose InO. Retrospective analyses of outcome determinants in these patients revealed low platelet count and high peripheral blast count as predictive of lack of response to InO.18 Promising data have recently been published about the use of InO in combination with low-intensity chemotherapy for newly diagnosed elderly patients with Ph− ALL.19 InO was given on day 3 of the first 4 cycles of multiagent chemotherapy, followed by a 3-year maintenance phase. Fifty-two patients (median age, 68 years) were treated. Although the accepted 2-year RFS for similar patients treated with chemotherapy alone is 20% to 30%, 2-year PFS for trial patients was 59% (median follow-up, 29 months). Overall, fractionated dosing of InO is thought to lead to less toxicity than single-dose schedules.
Pediatric experience with InO
The published pediatric experience with InO to date is limited to case reports and retrospective analyses. Bhojwani et al reported outcomes in a cohort of pediatric patients with heavily pretreated relapsed/refractory ALL who received InO through compassionate use agreements (51 patients treated at 30 centers in the United States, Australia, and Europe).20 The CR/CRh rate in this cohort was 67%: of these patients, 71% achieved MRD negativity. EFS and OS at 12 months for the entire cohort were 23.4% and 36.3%, respectively. Twenty-one patients in CR went on to HSCT. No baseline characteristics were found to predict response to InO in this cohort, but CD22 downregulation was noted in some cases of relapse. Toxicities were similar to those seen in the adult studies; of the 21 patients who went on to HSCT, 11 (52%) developed SOS.
With the high CD22 expression and prior encouraging response of this target in childhood ALL, the promising outcomes in adults treated with InO, and the need for additional therapeutic options in the event of CD19+ or CD19− relapse occurring following other CD19-directed therapy, the use of InO was targeted for pediatric development. COG AALL1621 is a phase 2 study of single-agent InO that is open and actively recruiting patients with second or greater relapse of CD22+ ALL and has met the initial recruitment goal of 48 patients to date. Although outcome data are not yet reported, feasibility has been demonstrated and stopping boundaries have not been exceeded. The Innovative Therapies for Childhood Cancer (ITCC) study 059 is testing a dose-escalation design of InO in the relapsed/refractory setting in a European multicenter study and will use a phased approach to test InO as a single agent prior to incorporating it into a modified UKALL R3 combination backbone. One goal of this study is to gather sufficient data to consider testing a substitution of InO instead of mitoxantrone for consideration in future high- and very high-risk ALL treatment. To date, the toxicities observed in both ITCC and COG pediatric studies are in line with those seen in adult patients with ALL, and the pharmacokinetics response, including MRD assessments and immune profiling of patients, is being tested in these ongoing studies (C. M. Zwaan and M. O’Brien, oral communications, July 2019).
With the ongoing pediatric InO studies in the relapsed/refractory setting, this agent is now being incorporated into the next frontline high-risk ALL study in the COG (study AALL1732) to test the hypothesis of whether 2 blocks of InO can be safely added to a modified augmented Berlin-Frankfurt-Muenster (BFM; modified BFM chemotherapy regimen [mBFM]) chemotherapy backbone in patients with high-risk B-ALL, and will improve the 5-year DFS compared with mBFM chemotherapy alone. Patients eligible for this study are estimated to have a current 5-year DFS of ∼65% to 86% based on recent COG and BFM study outcomes. Patients with high-risk B-lymphoblastic lymphoma and with mixed phenotype acute leukemia are eligible for this trial but will not receive InO and are not included in the primary statistical analysis. Planned enrollment will be 2269 children and young adults with high-risk B-ALL over ∼5 years, with the goal to improve their 5-year DFS to 87.8%. Other study objectives will include questions about equal duration of treatment of boys and girls, and for patients with mixed phenotype acute leukemias and high-risk B-lymphoblastic lymphoma. Figure 2 outlines the general schema for AALL1732.

Schematic diagram of COG study AALL1732 showing the randomization between patients receiving standard chemotherapy alone vs standard chemotherapy plus 2 blocks of InO after consolidation and MRD assessment. Schema shown with the permission of COG AALL1732 Study Chairs Jennifer McNeer and Maureen O’Brien.
Schematic diagram of COG study AALL1732 showing the randomization between patients receiving standard chemotherapy alone vs standard chemotherapy plus 2 blocks of InO after consolidation and MRD assessment. Schema shown with the permission of COG AALL1732 Study Chairs Jennifer McNeer and Maureen O’Brien.
Moving CAR-T therapy to the front line
CAR-T cells are T cells that are cultured ex vivo and genetically engineered to produce a chimeric receptor composed of an antigen-recognition domain from an antibody, a transmembrane domain, an intracellular signaling domain from CD3 ζ chain, and, in the case of second-generation CAR-T (which are those from which the bulk of clinical trial data have been derived), a costimulatory domain. They are intended to self-activate once the antigen-recognition domain binds its antigen on a target cell and to perform cytolytic functions in an major histocompatibility complex (MHC)-agnostic manner, bypassing the normal checks and balances of immune regulation. The first patient treated with CD19-directed CAR-T was an adult patient with refractory CLL who achieved CR after 28 days that was sustained for at least 10 months.21 This success was followed in parallel for children with ALL by studies at Children’s Hospital of Philadelphia, the Pediatric Oncology Branch at the National Cancer Institute, and Seattle Children’s Hospital for children with ALL; several pediatric phase 1 trials of different CD19 CAR-T products determined the maximum tolerated dose to be 1 million CAR-T per kilogram of body weight including different viral constructs and manufacturing sites.22-24 Multiple CAR-T products are in trial for childhood ALL and non-Hodgkin lymphoma globally now with a variety of vectors and approaches.
Lee et al reported a 70% CR rate and a 60% MRD− rate in 21 children and adults with relapsed/refractory ALL. All patients enrolled were included in the analysis, 8 of whom had prior HSCT.24 The authors noted that CAR-T can be an effective bridge to transplant for many patients who cannot otherwise achieve optimal reduction in disease burden to make HSCT effective. In a cohort study of 30 children and adults treated with the University of Pennsylvania anti-CD19 CAR-T product, CTL019,22 all patients had refractory B-lineage ALL. One month postinfusion, CR was observed in 27 patients who were able to receive product (90%), 2 of whom were refractory to blinatumomab; 15 had previously received allogeneic HSCT. Twenty-two of these patients (81%) were MRD− by flow cytometry. Six-month EFS and OS were 67% and 78%, respectively, although 7 responders relapsed at time points between 6 weeks and 8.5 months postinfusion and the analyses reported were by intent to treat; thus, not all patients were included in the outcomes reported. In building on sequential CAR-T product experience, Gardner et al refined the manufacturing procedures and reported the results of a phase 1 trial whereby the CAR-T product was designed to have consistent a CD4/CD8 1:1 ratio upon infusion, uniform CAR expression, and limited T-cell differentiation in culture.23 Forty-five children and young adults with relapsed/refractory ALL were infused with 93% efficacy in achieving the goal product parameters. Intent-to-treat analysis showed an 89% MRD− remission rate. The authors concluded that consistency of the CAR-T product enhanced disease response rates.
The phase 2 global trial of tisagenlecleucel25 screened 113 patients, enrolled 97, and final analysis reported outcomes for 75 pediatric and adult patients with CD19+ relapsed or refractory ALL who received the infused manufactured cell product. At 3 months, the CR/CRh rate was 81%, all of whom were also MRD− by flow cytometry. Twelve-month EFS and OS were 50% and 76%, respectively. Intent-to-treat analysis, which included those who did not receive an infusion due to disease progression or CAR-T manufacturing issues, resulted in a CR/CRh rate of 66%, more in line with the National Cancer Institute (NCI) study publication, suggests that heavily pretreated patients have multiple barriers to CAR-T infusion, but if infused, disease response rates can be high.
Early relapses after CAR-T therapy have tended to be CD19+ and due to lack of CAR-T persistence, whereas late relapses occur more often with CD19 antigen loss despite persistence of the CAR-T product. As such, many investigators are developing additional CAR-T products designed to target multiple antigens simultaneously, or with enhanced engineering technologies to help prevent antigen loss or resistance. Several examples including the PLAT series of trials at Seattle Children’s and third-party CARs developed at Memorial Sloan Kettering have taken these approaches to make products both more effective and/or more readily available to patients. Collaborations between academic and private-party pharmaceutical companies such as the NCI and Kite, the Fred Hutchinson Research Center, Memorial Sloan Kettering Cancer Center, and the Seattle Children’s Research Institute groups and Juno/Celgene, and the University of Pennsylvania and Novartis are examples of the mutually beneficial relationships that have advanced the science of CAR-T therapy to more widespread patient benefit.
These efforts also mark a rare instance in which an experimental agent has been tested on children and adults essentially simultaneously. Based on a series of promising results, the US FDA approved tisagenlecleucel, the first CAR-T commercial product for patients up to 25 years of age with B-cell precursor ALL that is refractory or in second or later relapse in August 2017. In October 2017, axicabtagene ciloleucel was approved for adult patients with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy, including diffuse large B-cell lymphoma not otherwise specified, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and diffuse large B-cell lymphoma arising from follicular lymphoma. Tisagenlecleucel received a subsequent approval in May 2018 for adults with relapsed or refractory large B-cell lymphoma. CAR-T cells grow and expand in the body leading to the potential for long-term persistence of an endogenous antileukemic or antilymphoma therapy. As such, CAR-T cells have been called “the first living drug” and, in reality, are the first gene/cell therapies for cancer that have attained regulatory approval.
A subset of children and adolescents with ALL still have a very poor prognosis despite the numerous advances made over decades. Risk-adapted therapy has improved, but not sufficiently for outcomes in patients who at diagnosis are known to have the highest-risk forms of ALL, including hypodiploid karyotype, TP53 mutations, t(17;19), infants with KMT2A-AFF4 fusion, iAMP21, and those who remain with molecularly or grossly positive residual leukemia after completion of induction and consolidation chemotherapy. These patients often relapse either on therapy or shortly after completing therapy and are often refractory to additional reinduction attempts or have a dismal salvage rate. HSCT is often difficult to realize for these patients because they may not attain a molecularly negative disease state to meet transplant criteria. The potential to activate the immune system to induce more effective remission has led investigators to consider the use of immunotherapy including CAR-T cells in several settings, including early in therapy before a patient has the chance to relapse. At the present time, over 90 CAR-T trials are available globally for patients with ALL targeting various antigens and using a variety of viral vectors and engineering strategies; however, only a small fraction are accessible to most patients due to eligibility restrictions, geographic distribution, manufacturing considerations, and financial challenges associated with reimbursement and payor coverage. Nonetheless, CAR-T products with a variety of promising targets including FLT3 for patients with FLT3+ acute myeloid leukemia or KMT2A-rearranged leukemias and the thymic stromal lymphopoietin protein receptor for patients with CRLF2+ (Ph-like) ALL are advancing toward availability through clinical trials.
Based on data using CAR-T cells in children, a proportion of children with very high-risk or highly refractory disease may experience long-term remission with this approach, and there is a suggestion that CAR-T cells may durably persist in patients, potentially eliminating the need for SCT and its consequent late effects in some patients with high-risk B-ALL. Given these factors, the COG has opened a limited institution phase 2 open-label study (AALL1721) for patients ages 1 to 25 years at initial diagnosis of CD19+ expressing B-ALL who have de novo NCI high-risk features and MRD ≥0.01% at the end of consolidation chemotherapy. Patients must have both adequate organ function and performance status and have received appropriate induction chemotherapy according to a standard multiagent regimen (specified in the study). Patients with Ph+ ALL, prior tyrosine kinase inhibitor therapy, those with hypodiploid ALL, or those with inherited bone marrow failure syndromes are not eligible. Also not eligible are patients with ≥25% blasts by morphology at the completion of induction chemotherapy or patients with ≥5% marrow blasts by morphology or persistent extramedullary disease at the end of consolidation chemotherapy. The objectives of this study are to estimate a variety of parameters of outcome and biologic end points with the primary goal to determine 5-year DFS in ∼140 patients treated with tisagenlecleucel. The schema for the CAR-T arm of AALL1721 is shown in Figure 3.

Schematic diagram of COG study AALL1721. The center column shown in gray shows the blocks of patient treatment. The left column shown in pink indicates the different phases of treatment and follow-up. The right column shown in green shows the specific components of the CAR-T product manufacturing process and how the timing of manufacturing parallels the treatment phases. Schema modeled after and shown with permission of COG AALL1721 Study Chair Shannon Maude.
Schematic diagram of COG study AALL1721. The center column shown in gray shows the blocks of patient treatment. The left column shown in pink indicates the different phases of treatment and follow-up. The right column shown in green shows the specific components of the CAR-T product manufacturing process and how the timing of manufacturing parallels the treatment phases. Schema modeled after and shown with permission of COG AALL1721 Study Chair Shannon Maude.
Summary
Landmark changes in the treatment of a wide variety of cancers were focused on immunotherapy over the past several years, and our knowledge is advancing exponentially. Initially, these treatments are all tested in patients with relapsed or refractory disease, often after many years of multiagent, immunosuppressive chemotherapy and sometimes following at least 1 stem cell transplant regimen. Thus, the patients in whom these agents are first used are often the least immunologically competent. The observation of good outcomes for some patients with single-agent immunotherapies is quite impressive and leads to the conjecture that if such good outcomes can be seen for patients with advanced and refractory disease, treating patients earlier in their disease course could yield even better results. Immunotherapeutic agents have distinct mechanisms of action and toxicity profiles compared with conventional cytotoxic chemotherapy agents. Some toxicities of immunotherapies are shared, and the most significant are on-target and related to the mechanism of action of the treatment. In an attempt to continue to improve outcomes for patients with pediatric ALL and to reduce toxicities, both short- and long-term, exploring the introduction of immunotherapies earlier in treatment of children with ALL lays important groundwork for potentially decreasing the burdens of conventional therapy and even potentially shortening the duration of treatment. Although these new therapies carry a high cost, the potential to reduce cardiac, endocrine, neuropsychologic, orthopedic, ocular, dental, and other toxicities for survivors may carry both a quality of life and a future financial benefit to individuals and to society as long as good outcomes are not compromised.
Current use of immunotherapies in pediatric ALL has advanced more rapidly for B-lineage disease, although more options are developing for T-lineage disease and with multitargeted strategies such as bicistronic and bispecific CARs and trispecific T-cell engager constructs among many others. Depending on the results of the trials testing blinatumomab, InO, and CAR-T products in the front line, it is possible that a generation of trials would explore reduction of cytotoxic chemotherapy in a substitution strategy to further reduce the risks of long-term side effects of traditional agents. Additional immunotherapy combination studies will continue to expand. Ultimately, the goal of therapy for all childhood cancers is to maximize survival with high quality of life and decrease frequencies and morbidities of toxicity for survivors, their families, and their communities at large. The first step toward this is to evaluate carefully the inclusion of these and other promising therapies in appropriately conducted and controlled trials to maximize knowledge and share data globally in order to better treatment strategies for all.
Acknowledgments
The authors thank the following colleagues for their contributions and collegiality in the preparation of this manuscript and the clinical trials described here: Anne Angiolillo, Patrick Brown, Stacy Cooper, Sumit Gupta, Stephen Hunger, Franco Locatelli, Mignon Loh, Shannon Maude, Jennifer McNeer, Maureen O’Brien, Elizabeth Raetz, Rachel Rau, Arend von Stackelberg, and C. Michel Zwaan.
Correspondence
Lia Gore, Children’s Hospital Colorado, 13123 East 16th Ave, Box B115, Aurora CO 80045; e-mail: lia.gore@cuanschutz.edu.
