
Abstract
Recent advances in our understanding of iron metabolism regulation and crosstalk with erythropoiesis have provided insight into the pathophysiology of multiple disease conditions. For instance, the peptide hormone hepcidin is central to the regulation of iron metabolism. Its effect on cellular iron concentration involves binding ferroportin, the main iron export protein, resulting in its internalization and degradation and leading to iron sequestration within ferroportin-expressing cells. Furthermore, hepcidin regulation by erythropoiesis is attributed in large part to a bone marrow–derived hormone erythroferrone. Erythroferrone-induced hepcidin suppression in diseases of expanded hematopoiesis results in iron overload. Conversely, diseases, such as iron refractory iron deficiency anemia and anemia of chronic inflammation, are characterized by aberrantly increased hepcidin, resulting in iron sequestration and decreased circulating iron and eventually leading to iron-restricted erythropoiesis. Lastly, because iron functions in concert with erythropoietin to promote erythroid precursor survival, proliferation, and differentiation, iron deficiency anemia is a consequence not only of decreased hemoglobin synthesis in each cell but also, a decrease in erythropoietin responsiveness in the bone marrow. How to translate this new information to the clinical setting has not been fully elucidated. The purpose of this manuscript is to summarize current standard tools for identifying iron deficiency in anemic patients; explore the tools and context for evaluating novel markers, such as hepcidin, erythroferrone, and markers of the iron restriction response; and assess available evidence for how their use could increase our understanding of health outcomes in clinically challenging cases.
Learning Objectives
Understand mechanisms involved in iron regulation and crosstalk with erythropoiesis
Review current standard diagnosis of iron deficiency, iron deficiency anemia, and determination of therapeutic approach
Evaluate the utility of novel markers of iron metabolism and erythropoiesis to further delineate clinical approach in multiple disease states
Case presentation
A 35-year-old female with medical history significant for hypothyroidism is referred to hematology for evaluation of anemia. Her hypothyroidism was diagnosed 12 years prior when she presented with fatigue and weight gain. She has been on a stable dose of synthroid for many years and presents having recently seen her endocrinologist for recurrence of fatigue. She complains of generalized weakness worsening over the last few months. Her thyroid function testing revealed well-controlled hypothyroidism. On further questioning, she reports more excessive bleeding during her menses in the last 6 months. She was also recently discharged from the local hospital after an admission for shortness of breath and cough, was diagnosed with pneumonia, and recently completed a course of antibiotics. Her cough and shortness of breath have resolved, but the fatigue and weakness persist. She denies evidence of PICA and has been told to eat more iron-rich foods with orange juice. Her hemoglobin (Hb) on the day of consultation is 10.8 g/dL, a fall from 12.6 g/dL 6 months ago, and she has normal mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH).
On the day of consultation, her physical examination was normal; thyromegaly, splenomegaly, evidence of chronic iron deficiency changes (eg, nail spooning, hair loss, glossitis, and cheilosis), or signs of infection were not noted. Thyroid function was repeated, and a full evaluation of anemia was performed. She again had a normal thyrotropin and no evidence of hemolysis (eg, normal lactate dehydrogenase, haptoglobin, and reticulocyte count). Her vitamin B12 and folate concentrations were normal. There was no evidence of renal or liver disease. Her transferrin saturation was 14%, and ferritin was 88 μg/L. Peripheral smear reveal normocytic normochromic red blood cells (RBCs) without toxic granulations or Dohle bodies and no evidence of hypersegmented neutrophils. Some poikilocytosis and anisocytosis were present, but schistocytes, pencil cells, and target cells were not seen.
Question
Would this anemic patient benefit from iron supplementation, and if so, is there an expectation that oral iron supplementation would work or is the parenteral route is preferable to both relieve symptoms and normalize Hb?
Introduction
Anemia is designated by a somewhat arbitrary method using Hb from healthy individuals, in which “normal” is defined as within the 95% around the mean value. Thus, individuals with values that fall in the lower 2.5% are diagnosed with anemia,1 and the definition of anemia may differ depending on patient gender, race, and age. In addition, understanding the timeline of Hb change is critical for determining the types of underlying etiologies to consider. Iron deficiency anemia and anemia of chronic inflammation (ACI) are the most common causes of anemia.2,3 Although iron deficiency anemia is commonly microcytic and hypochromic, ACI more typically ranges from normocytic to microcytic and normochromic to hypochromic. Increasing evidence more recently reveals that iron deficiency anemia is not universally microcytic, especially in elderly patients with compound causes of anemia, making for more complicated distinction between iron deficiency anemia and ACI. Both iron deficiency anemia and ACI present with decreased circulating serum iron concentration and transferrin saturation, but whereas iron deficiency anemia is characterized by depleted iron stores (ie, serum ferritin below the lower limit of normal), iron stores are ample in ACI. In the setting of inflammation, ACI may be difficult to differentiate from iron deficiency anemia, and iron deficiency anemia may coexist with ACI. In the hospital, this combination may be even more frequent, and it can be more difficult to identify the underlying cause in light of the multiple conditions, medications, and procedures that can induce sometimes transient inflammation and multiple and frequent diagnostic blood draws contributing to iron deficiency anemia. Furthermore, in rare cases, severe microcytic anemia results from iron refractory iron deficiency anemia (IRIDA), in which a mutation in the TMPRSS6 gene (encoding matriptase-2) leads to aberrant hepcidin expression and results in inadequate iron absorption and persistent iron sequestration.4 Such cases respond almost exclusively to parenteral iron. The main purpose of this manuscript is to review current consensus and standard practice in diagnosis of iron deficiency and iron-restricted erythropoiesis; explore whether novel diagnostic tools can further improve our ability to identify patients who would benefit from iron supplementation, even in the setting of inflammation; and provide predictive guidance on whether oral or parenteral iron administration is the preferable first-line approach to ameliorate iron deficiency and anemia.
Pathophysiology and regulation of iron metabolism
Because most of the body’s iron is destined for Hb synthesis, erythropoiesis and iron metabolism are inextricably linked. Iron must be tightly regulated to avoid shortfalls or excesses, which may result in anemia or iron overload, respectively. The stable concentration of circulating iron enables erythropoiesis and other iron-requiring physiological processes and is maintained by dietary absorption, storage, and recycling of iron. The peptide hormone hepcidin, secreted primarily by hepatocytes, is the principal regulator of iron homeostasis.5 Hepcidin downregulates iron release into the plasma by binding to and functionally downregulating ferroportin, the sole cellular iron exporter. The presence of ferroportin in duodenal enterocytes, splenic and liver macrophages, and hepatocytes (sites of iron absorption, recycling, and storage, respectively) enables hepcidin to regulate these functions in iron metabolism via its interaction with ferroportin.
Hepcidin regulates iron absorption by targeting ferroportin at the basolateral enterocyte, controlling the entry of iron into the circulation. However, iron must first enter at the apical side of the duodenal enterocyte via DMT1 (Figure 1). Thus, if increased iron requirements do not suppress hepcidin, iron taken up by DMT1 will be shed in the gastrointestinal tract as part of normal cellular turnover. However, when hepcidin levels are low, the presence of ferroportin on the basolateral surface enables iron egress from duodenal enterocytes, exporting iron into the circulation for transferrin binding (Figure 1). Controlling iron absorption in this manner may enable accumulation of iron within enterocytes during a meal and provide a longer window of opportunity to absorb iron than if regulation of iron absorption occurred at the apical surface. Lastly, hepcidin-mediated changes in enterocyte iron content also modulate the activity of the transcription factor HIF2α in enterocytes.6 Low levels of hepcidin during iron deficiency allow iron export from enterocytes, depletion of their cellular iron, inactivation of prolyl hydroxylase enzymes, and stabilization of HIF2α. HIF2α, in turn, increases transcription of DMT1, Dcytb, and ferroportin mRNA, coordinating the increase in both apical and basolateral iron transport.

Regulating iron absorption dependent on apical import and basolateral export. At the duodenal enterocyte, where iron is absorbed, hepcidin functions as a negative regulator at the basolateral surface, where ferroportin exports iron from duodenal enterocytes into the circulation. Hepcidin-mediated changes in enterocyte iron content modulate HIF2α in enterocytes. HIF2α is stabilized when hepcidin is low during iron deficiency, resulting in increased transcription of DMT1 and Dcytb as well as ferroportin messenger RNA Together, stabilization of HIF2α in the gastrointestinal tract with suppression of hepcidin would provide the most potent stimulus for iron absorption. In high-hepcidin states, ferroportin internalization and degradation result in the accumulation of iron within the enterocyte, and it is excreted in the stool with the normal frequent turnover of the intestinal mucosa. Dcytb, duodenal cytochrome B; DMT1, divalent metal transporter 1; Fe, iron; FPN1, ferroportin 1; Tf, transferrin.
Regulating iron absorption dependent on apical import and basolateral export. At the duodenal enterocyte, where iron is absorbed, hepcidin functions as a negative regulator at the basolateral surface, where ferroportin exports iron from duodenal enterocytes into the circulation. Hepcidin-mediated changes in enterocyte iron content modulate HIF2α in enterocytes. HIF2α is stabilized when hepcidin is low during iron deficiency, resulting in increased transcription of DMT1 and Dcytb as well as ferroportin messenger RNA Together, stabilization of HIF2α in the gastrointestinal tract with suppression of hepcidin would provide the most potent stimulus for iron absorption. In high-hepcidin states, ferroportin internalization and degradation result in the accumulation of iron within the enterocyte, and it is excreted in the stool with the normal frequent turnover of the intestinal mucosa. Dcytb, duodenal cytochrome B; DMT1, divalent metal transporter 1; Fe, iron; FPN1, ferroportin 1; Tf, transferrin.
Hepcidin transcription itself is increased by iron loading and elevated levels of inflammatory cytokines, but it is suppressed by tissue hypoxia, iron deficiency, and ineffective erythropoiesis (Figures 2 and 3). Regulation of hepcidin by local iron stores involves the bone morphogenic protein (BMP) pathway, specifically BMP6 and BMP2,7 with hemojuvelin (HJV) as an iron-specific BMP coreceptor and stimulant of the BMP type I receptor in iron overload states.7 Regulation of hepcidin by systemic iron concentration involves iron transferrin binding transferrin receptor 1 (TfR1) and TfR2 in the liver. Iron binding TfR1 displaces high iron (HFE), and binding to TfR2 stabilizes the receptor. HFE and TfR2 then likely interact with HJV and potentiate the BMP pathway signaling via phosphorylation of SMAD1/5/8, increasing hepcidin transcription in response to iron7,8 (Figure 2). Hepcidin induction during inflammation is mediated by interleukin-6,9 signaling via the STAT3 pathway in hepatocytes. Both pathways inducing hepcidin result in iron sequestration within duodenal enterocytes and iron-recycling macrophages, hypoferremia, and restricted iron availability for erythropoiesis (Figure 3).
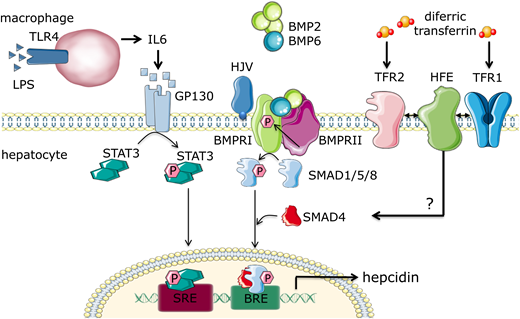
Multifactorial regulation of hepcidin expression in hepatocytes. Hepcidin is regulated via multiple pathways. In inflammatory states, macrophages release interleukin-6 (IL-6), which activates the STAT3 pathway in hepatocytes, leading to production of hepcidin. Local iron stores induce nonhepatocyte liver cells to produce bone morphogenic protein 2 (BMP2) and BMP6, which stimulate BMP receptor I; this activates the s-mothers against decapentaplegic 1/5/8 (SMAD1/5/8) pathway, which in concert with SMAD4, translocates to the nucleus to induce hepcidin transcription. Increased systemic iron leads to presence of diferric transferrin compounds, which bind TfR1, disrupting its interaction with HFE, HFE complexing with TfR2, BMP receptor, or hemojuvelin (HJV), an iron-specific BMP coreceptor; this leads to potentiation of the BMP/SMAD1/5/8 signaling pathway and hepcidin transcription. BRE, TFIIB recognition element; LPS, lipopolysaccharide; SRE, serum response element.
Multifactorial regulation of hepcidin expression in hepatocytes. Hepcidin is regulated via multiple pathways. In inflammatory states, macrophages release interleukin-6 (IL-6), which activates the STAT3 pathway in hepatocytes, leading to production of hepcidin. Local iron stores induce nonhepatocyte liver cells to produce bone morphogenic protein 2 (BMP2) and BMP6, which stimulate BMP receptor I; this activates the s-mothers against decapentaplegic 1/5/8 (SMAD1/5/8) pathway, which in concert with SMAD4, translocates to the nucleus to induce hepcidin transcription. Increased systemic iron leads to presence of diferric transferrin compounds, which bind TfR1, disrupting its interaction with HFE, HFE complexing with TfR2, BMP receptor, or hemojuvelin (HJV), an iron-specific BMP coreceptor; this leads to potentiation of the BMP/SMAD1/5/8 signaling pathway and hepcidin transcription. BRE, TFIIB recognition element; LPS, lipopolysaccharide; SRE, serum response element.

Systemic regulation of iron metabolism and utilization. Iron is either absorbed in the duodenum or recycled from senescent RBCs, and it is found in circulation bound to transferrin (diferric transferrin complex). Diferric transferrin delivers iron to the bone marrow for Hb synthesis in erythroid precursors. Erythropoiesis also requires the presence of erythropoietin (Epo) to enable erythroblast differentiation and enucleation. Epo-stimulated erythroblasts secrete erythroferrone (ERFE), which functions by sequestering bone morphogenic protein 6 (BMP6) in circulation and decreasing signaling to and suppression of hepcidin. Hepcidin functions in multiple ways to decrease circulating iron, including blocking release of iron from splenic macrophages and preventing iron absorption in the gut. This interrelated process is intended to prevent shortfalls and excess of systemic iron and fine tune iron transport to where it is needed. FPN1, ferroportin 1. Fe, iron; Tf, transferrin.
Systemic regulation of iron metabolism and utilization. Iron is either absorbed in the duodenum or recycled from senescent RBCs, and it is found in circulation bound to transferrin (diferric transferrin complex). Diferric transferrin delivers iron to the bone marrow for Hb synthesis in erythroid precursors. Erythropoiesis also requires the presence of erythropoietin (Epo) to enable erythroblast differentiation and enucleation. Epo-stimulated erythroblasts secrete erythroferrone (ERFE), which functions by sequestering bone morphogenic protein 6 (BMP6) in circulation and decreasing signaling to and suppression of hepcidin. Hepcidin functions in multiple ways to decrease circulating iron, including blocking release of iron from splenic macrophages and preventing iron absorption in the gut. This interrelated process is intended to prevent shortfalls and excess of systemic iron and fine tune iron transport to where it is needed. FPN1, ferroportin 1. Fe, iron; Tf, transferrin.
Erythroid regulation of hepcidin
Regulation of hepcidin by erythropoiesis comes from observations that disorders of ineffective erythropoiesis (eg, β-thalassemia, myelodysplastic syndromes, and dyserythropoietic anemias) exhibit lower than expected hepcidin expression, despite iron overload. In fact, suboptimal hepcidin expression is implicated in iron overload in these diseases and supports the erythropoietic signal on hepcidin regulation.10,11 Convincing evidence suggests that erythroid suppression of hepcidin is a direct consequence of increased erythropoietic activity itself, irrespective of anemia, hypoxia, and increased erythropoietin (Epo).12 Such an “erythroid factor” must be secreted by erythroid precursors, functioning as a hormone to suppress hepcidin expression in the liver (Figure 3). A potential physiological regulator of hepcidin, erythroferrone (ERFE), has been identified.13 Supporting evidence reveals loss of hepcidin suppression after phlebotomy in Erfe knockout mice, increased Erfe in mouse models of β-thalassemia, and relatively increased hepcidin expression and decreased iron overload in β-thalassemic/Erfe knockout relative to β-thalassemic mice.14 Most recently, a mechanism of ERFE regulation of hepcidin has been proposed, providing evidence of BMP6 sequestration by ERFE and resulting in a BMP/SMAD-dependent decrease in signaling to hepcidin expression.15
The discoveries of hepcidin as a central regulator of iron metabolism and erythroid regulation of hepcidin by ERFE have enabled a more comprehensive exploration of aberrant iron metabolism and molecular mechanisms underlying this effect in many clinical scenarios. Specifically, the predictive value of iron stores and erythropoietic iron responsiveness to support the management of anemias has not yet been fully explored.
Molecular mechanisms underlying the iron restriction response
Because iron functions in concert with Epo to promote erythroid precursor survival, proliferation, and differentiation, the “iron restriction response” serves as the basis for decreased bone marrow Epo sensitivity and repression of erythropoiesis, resulting in anemia during iron deficiency. Thus, iron restriction because of systemic depletion of iron stores as well as functional iron deficiency in high hepcidin states (eg, during inflammation from multiple causes or IRIDA) serves as a brake on erythropoiesis when iron availability is limited (Figure 4A). Mechanisms underlying the iron restriction response are unresolved but likely involve specialized regulators. For example, although iron restriction impairs Epo responsiveness, it does so selectively, preserving signaling to erythroid precursor survival and proliferation without inducing differentiation.16 First, iron restriction results in the loss of iron-sulfur cluster, inactivating aconitases in erythroblasts; recent evidence demonstrates that aconitase inactivation results in decreased Epo responsiveness in vitro and anemia in vivo. Second, TfR2 is involved in transferrin-iron trafficking, EpoR trafficking to the membrane, and potentially, communicating extracellular iron availability to mitochondrial aconitase.17 In support of this are experiments using TfR2 knockout transplanted mice with loss of TfR2 only in the bone marrow; these mice exhibit inappropriately increased erythroid expansion in response to iron deficiency,18 suggesting the importance of TfR2 in the iron restriction response. Last, SCRIBBLE, a master regulator of receptor trafficking and signaling, is involved in EpoR surface concentration, regulated by TfR2, and essential for erythroid iron restriction response.19 Taken together, iron restriction functions via targeted erythroid precursor sensitivity to inflammation in an Epo-independent manner, involving aconitase, TfR2, and SCRIBBLE to modify EpoR-dependent signaling to support cell viability and proliferation in the absence of differentiation (Figure 4A-B). More recent evidence also suggests that iron deficiency results in G1 arrest owing to the requirement of iron for ribonucleotide reductase during DNA replication.20 Mechanisms underlying this effect involve nuclear receptor coactivator 4 (NCOA4), an important cargo receptor necessary for the extraction of iron from ferritin, which aids in the coordination of iron availability and erythroid differentiation to protect cells from replication stress21,22 (Figure 4C). Exploring coordination of parameters involved in the iron restriction response may provide novel therapeutic targets for patients with ACI and possibly, other diseases.
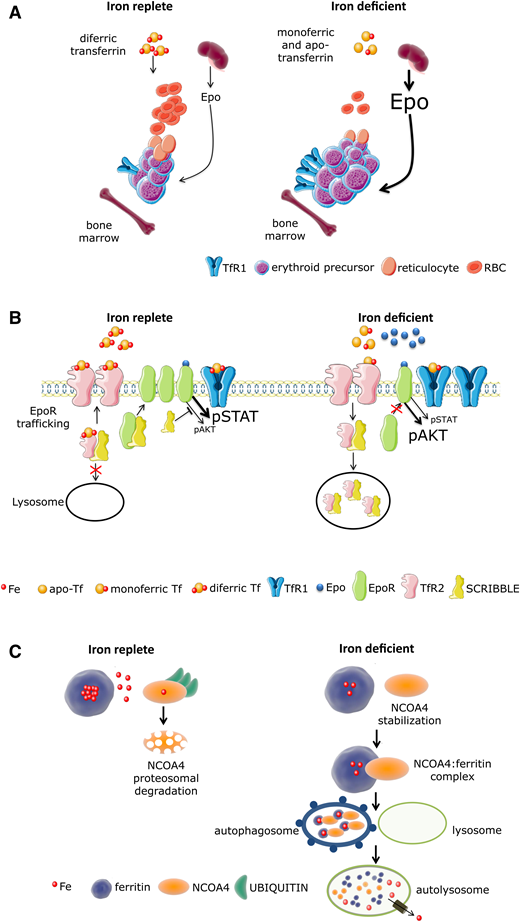
Model of iron regulation in replete and restricted erythropoiesis. (A) In the iron-replete condition, small amounts of Epo induce proliferation and differentiation of erythroblasts that take up iron by binding diferric transferrin at TfR1 for Hb synthesis. During iron deficiency, circulating transferrin is either devoid of iron (apotransferrin) or bound to iron at only 1 binding site (monoferric transferrin) with a significantly decreased concentration of diferric transferrin. Thus, in addition to less iron uptake owing to a decrease in diferric transferrin and resulting in a decrease in Hb synthesis per cell, iron restriction decreases Epo responsiveness, modifying Epo receptor–dependent signaling to support cell viability in excess of erythroid proliferation and differentiation. (B) In iron-replete conditions, diferric Tf binding to TfR2 prevents trafficking of TfR2-associated vesicles to lysosomes, enabling “free” SCRIBBLE to promote efficient EpoR surface presentation and normal Epo responsiveness. In iron-deficient conditions, TfR2-associated vesicles traffic predominantly to lysosomes, accelerating catabolism of TfR2-Scribble complexes and resulting in impaired Scribble-mediated EpoR delivery to the cell surface. Because SCRIBBLE suppresses AKT signaling, STAT5-dependent signal predominates in iron-replete conditions, supporting erythroid differentiation. In contrast, trafficking of Scribble to the lysosome in iron-deficient conditions results in persistent AKT signaling, impeding erythroid differentiation. (C) Nuclear receptor coactivator 4 (NCOA4) is a selective cargo receptor for autophagic ferritin turnover, critical for regulation of intracellular iron availability. In iron-replete states, NCOA4 binds iron, undergoes ubiquitination, and is targeted to the proteasome for degradation. In iron-deficient conditions, NCOA4 is stabilized and participates in trafficking ferritin to the lysosome, enabling iron release from ferritin and transport to the mitochondria for heme synthesis. This orchestration of erythropoiesis in parallel with iron availability prevents the proliferation and differentiation of erythroblasts when there is insufficient iron to support commensurate Hb synthesis and protects from replication stress in the absence of adequate iron. The mechanisms involved are beginning to come to light. AKT, protein kinase B; EPO, erythropoietin; EpoR, erythropoietin receptor; Fe, iron; NCOA4, nuclear receptor coactivator 4; RBC, red blood cell; STAT, signal transducer and activator of transcription; Tf, transferrin; TfR, transferrin receptor.
Model of iron regulation in replete and restricted erythropoiesis. (A) In the iron-replete condition, small amounts of Epo induce proliferation and differentiation of erythroblasts that take up iron by binding diferric transferrin at TfR1 for Hb synthesis. During iron deficiency, circulating transferrin is either devoid of iron (apotransferrin) or bound to iron at only 1 binding site (monoferric transferrin) with a significantly decreased concentration of diferric transferrin. Thus, in addition to less iron uptake owing to a decrease in diferric transferrin and resulting in a decrease in Hb synthesis per cell, iron restriction decreases Epo responsiveness, modifying Epo receptor–dependent signaling to support cell viability in excess of erythroid proliferation and differentiation. (B) In iron-replete conditions, diferric Tf binding to TfR2 prevents trafficking of TfR2-associated vesicles to lysosomes, enabling “free” SCRIBBLE to promote efficient EpoR surface presentation and normal Epo responsiveness. In iron-deficient conditions, TfR2-associated vesicles traffic predominantly to lysosomes, accelerating catabolism of TfR2-Scribble complexes and resulting in impaired Scribble-mediated EpoR delivery to the cell surface. Because SCRIBBLE suppresses AKT signaling, STAT5-dependent signal predominates in iron-replete conditions, supporting erythroid differentiation. In contrast, trafficking of Scribble to the lysosome in iron-deficient conditions results in persistent AKT signaling, impeding erythroid differentiation. (C) Nuclear receptor coactivator 4 (NCOA4) is a selective cargo receptor for autophagic ferritin turnover, critical for regulation of intracellular iron availability. In iron-replete states, NCOA4 binds iron, undergoes ubiquitination, and is targeted to the proteasome for degradation. In iron-deficient conditions, NCOA4 is stabilized and participates in trafficking ferritin to the lysosome, enabling iron release from ferritin and transport to the mitochondria for heme synthesis. This orchestration of erythropoiesis in parallel with iron availability prevents the proliferation and differentiation of erythroblasts when there is insufficient iron to support commensurate Hb synthesis and protects from replication stress in the absence of adequate iron. The mechanisms involved are beginning to come to light. AKT, protein kinase B; EPO, erythropoietin; EpoR, erythropoietin receptor; Fe, iron; NCOA4, nuclear receptor coactivator 4; RBC, red blood cell; STAT, signal transducer and activator of transcription; Tf, transferrin; TfR, transferrin receptor.
Current standard approach to delineating iron status in anemia
Assessing the underlying etiology of anemia has classically involved an algorithm to identify decreased production (hypoproliferative) or increased destruction (hemolytic) of RBCs. Truly hypoproliferative anemia is a consequence of decreased stem cell number and/or differentiation, typically as a consequence of parvovirus B19, bone marrow infiltration, immune-mediated processes (eg, aplastic anemia), or decreased Epo production (eg, renal failure). Disordered erythroblast differentiation also results in a hypoproliferative hematopoiesis and is caused by defective nuclear (eg, vitamin B12 or folate deficiency) or cytoplasmic (eg, iron deficiency) maturation owing to limited DNA or Hb synthesis, respectively. Hemolytic anemia, however, is a consequence of imperfect bone marrow compensation in the setting of a higher rate of circulating RBC destruction. Reticulocytosis is a typically used surrogate measure of bone marrow responsiveness such that hypoproliferative anemias have relatively low or normal reticulocyte counts, whereas hemolytic anemias or conditions associated with increased extramedullary destruction are typically associated with compensatory increases in hematopoiesis as reflected by increased reticulocyte counts. Several nomograms are available to quantify the daily production of reticulocytes specifically to address this question. The best cited is the reticulocyte production index (RPI), which correlates the hematocrit with the expected earlier release of bone marrow reticulocytes and consequent number of days that these early reticulocytes can be found in the circulation.23 Thus, decreased RPI (<2) would be expected in hypoproliferative anemias (Figure 5); whether the underlying cause is systemic iron deficiency, iron-restricted erythropoiesis, or other would depend on an array of additional parameters (eg, MCV, MCH, peripheral smear, serum ferritin, iron, transferrin concentration, and transferrin saturation).

Algorithm for diagnosis of iron deficiency anemia and ACI using clinically available tools. *In certain circumstances, patients with ferritin >100 μg/L may be responsive to iron supplementation (eg, chronic renal insufficiency and congestive heart failure). CHr, reticulocyte corpuscular hemoglobin; F, female; HYPO%, percent hypochromic red blood cells; M, male; sTfR1, soluble transferrin receptor 1; Tf sat, transferrin saturation.
Algorithm for diagnosis of iron deficiency anemia and ACI using clinically available tools. *In certain circumstances, patients with ferritin >100 μg/L may be responsive to iron supplementation (eg, chronic renal insufficiency and congestive heart failure). CHr, reticulocyte corpuscular hemoglobin; F, female; HYPO%, percent hypochromic red blood cells; M, male; sTfR1, soluble transferrin receptor 1; Tf sat, transferrin saturation.
Ferritin is the main intracellular iron storage protein; serum ferritin is thus a surrogate indirect measure of iron stores. Its shortcomings result from its additional function as an acute-phase reactant in response to inflammation, complicating the assessment of iron stores during infections and inflammation. Serum iron is influenced by the content and timing of meals and the dynamic movement of iron between compartments (ie, balance between iron uptake by and export out of cells). Transferrin is the main iron carrier protein in circulation, and its saturation is a calculated measure dependent both on its concentration (ie, production by the liver) and the amount of iron present in the circulation. In addition, activation of the immune and inflammatory responses results in increased production of inflammatory markers (eg, C-reactive protein [CRP]), which can be used to corroborate clinical and other laboratory evidence of inflammation. Thus, transferrin saturation <20% or serum ferritin <20 μg/L is consistent with iron deficiency anemia, and serum ferritin >100 μg/L is typically consistent with ACI (Figure 5). These standard metrics are useful in distinguishing between systemic iron deficiency and ACI in the majority of cases, guiding clinical decision making toward the potential benefit from iron supplementation when iron deficiency is the likely cause of anemia.
Iron deficiency is also estimated to affect large fractions of patients with chronic heart failure, chronic kidney disease, and inflammatory bowel disease, all chronic illnesses with significant inflammatory components. Thus, although we use serum ferritin >100 μg/L to delineate the cutoff above which iron supplementation is considered ineffective, there is increasing compelling evidence that, at least in some chronic illnesses, patients with ferritin 100 to 300 μg/L respond to parenteral iron, with increased Hb when anemia coexists,24-26 in addition to improvement in cardiac function in patients with chronic heart failure.25,26 Similarly, studies using ferritin cutoffs in excess of 100 μg/L indicate that iron supplementation is effective in treating anemia in chronic kidney disease27 and the oncological setting,28 even when ferritin is 300 to 500 μg/L in some studies.
Additional diagnostic tools enable evaluation of iron status and predict potential responsiveness to iron supplementation in cases where transferrin saturation and serum ferritin values are intermediate between clear iron deficiency and ACI (Figure 5). In such situations, serum soluble transferrin receptor 1 (sTfR1) has been proposed to quantify “iron avidity” of erythroblasts and reticulocytes in the bone marrow.29 Although TfR1 is critical for uptake of transferrin-bound iron in all cells, erythroblasts and reticulocytes express TfR1 at higher concentration than any other cell type. This is the consequence of high iron requirements for Hb synthesis. TfR1 undergoes posttranscriptional modification during iron deficiency, resulting in the potential to increase uptake of transferrin-bound iron. Furthermore, because Epo stimulates TfR1 expression and Epo levels are increased during iron deficiency, erythroblast TfR1 expression is further increased in conditions of iron-restricted erythropoiesis (Figure 4A). Serum sTfR1 is the truncated extracellular fraction of the membrane-bound TfR130 shed from reticulocytes inside exosomes, undergoing cleavage off exosomes to the form found in circulation; serum sTfR1 is proportional in concentration to membrane-bound TfR1 and thus, reflects both the number and the iron status of erythroid precursors.
Multiple enzyme-linked immunosorbent assays (ELISAs), immunoenzymatic assays, latex-enhances immunoassays, and fluoroimmunoassays are available for clinical assessment of sTfR1. The latest versions of these quantitative assays are sensitive, precise, and compatible with automated systems. Lack of standardization, however, remains an issue, with different assays using different standards, reference ranges, and units (ie, grams per liter and nanomoles per liter). Despite this and because sTfR1 concentration is unaffected by inflammation, these assays, by enabling indexes (such as sTfR/log ferritin), have assisted in the clinical determination of concurrent systemic iron deficiency with ACI (Figure 5). A relatively increased sTfR1 suggests that the erythron is iron deficient and avid even in the setting of ACI when elevated hepcidin and iron sequestration result in decreased iron availability for erythropoiesis, raising the possibility that such patients may respond to iron supplementation.
Lastly, reticulocyte corpuscular hemoglobin (CHr), percent hypochromic red blood cells (HYPO%), and zinc protoporphyrin-to-heme ratio (ZPP/H) have been identified as markers that dynamically reflect iron availability during recent Hb synthesis.31-34 Decreased CHr (ie, <28 pg) and increased HYPO% (ie, >5%) contribute significantly to the algorithm identifying a component of iron-restricted erythropoiesis in both anemia and nonanemic patients with normal serum ferritin levels.31-33 Specifically, a combination of markers (ie, transferrin saturation, HYPO%, CHr, and sTfR1) identified iron-restricted erythropoiesis in 36% of patients with ferritin levels 31 to 100 μg/L and 16% of patients with ferritin levels 101 to 300 μg/L31 (Figure 5). However, the sensitivity and specificity of CHr or HYPO% as individual markers to identify iron-restricted erythropoiesis are relatively low in nonanemic patients, because they are likely only affected when erythropoietic activity is enhanced, namely during stress erythropoiesis.31 These findings warrant independent verification using a gold standard confirmation of iron-restricted erythropoiesis. Increased ZPP/H formation (as a reflection of iron-zinc competition for binding ferrochelatase, the last step in Hb synthesis) is a well-known biomarker with established reference ranges and potential as a screening test for iron deficiency in the clinical setting. Recent data demonstrate that ZPP/H values >107 and >90 μmol/mol heme have high sensitivity and specificity and are predictive of iron deficiency in healthy community-dwelling Indian women and preschool children, respectively.32 Because increased ZPP/H does not distinguish between systemic iron deficiency and iron deficiency erythropoiesis, serum ferritin is subsequently needed to assess whether oral or parenteral iron supplementation is most useful, but its ease of use underscores its viability as a screening test to, when normal, exclude iron deficiency as a cause of anemia.
Taken together, detecting iron deficiency (which is instrumental to prescribing iron supplementation) in patients with concomitant overt or subclinical inflammation when ferritin levels are >100 μg/L remains an unresolved issue. The unmet clinical need for defining iron deficiency when ferritin is >100 μg/L owing to concurrent inflammation may benefit from a more nuanced assessment, in a nonbinary manner, to provide insight into both the regulation and compensation required to coordinate iron requirements for erythropoiesis. Likely, hepcidin levels can assist particularly when hepcidin assays become standardized and reference ranges become available. A recent elegant study using hepcidin in humans confirms that the suppressing signal by iron deficiency overrides the stimulation by inflammation, suggesting that hepcidin can identify iron deficiency and predict responsiveness to oral iron supplementation in more complex settings.33
Potential utility of novel markers
Hepcidin, ERFE, and markers of the iron restriction response may be additionally useful in complex cases. For example, hepcidin may be a useful diagnostic tool in predicting response to oral iron supplementation in IDA, in diagnosis of iron overload disorders, and as a marker of inflammation as the cause of anemia (ie, ACI). Whether the utility of this metric exceeds currently available tools or whether the information that it provides significantly contributes to further the currently available approaches (ie, serum ferritin, transferrin saturation, sTfR1, CHr, ZPP/H, and markers of nonspecific inflammation; eg, CRP) will require its application in clinical research settings to discern. To date, several studies explore this question.
One manuscript introduced the “Thomas Index,” which uses hepcidin 4 nmol/L as the cutoff to distinguish iron deficiency anemia from ACI with concurrent iron deficiency anemia and CHr to further discriminate between ACI and ACI with concurrent iron deficiency anemia.34,35 The authors argue that the utility of hepcidin relative to ferritin can be attributed to its more immediate representation of iron availability for erythropoiesis. A multicenter observational prospective cohort study of >1000 patients evaluated serum hepcidin <20 ng/L as a marker of iron deficiency; this study identified 37% of patients with evidence of iron deficiency (relative to 6% identified by serum ferritin <100 μg/L and 13% identified by sTfR1/log ferritin >0.8) and predicted increased 1-year mortality (adjusted odds ratio [OR], 1.51) as well as poor physical recovery (adjusted OR, 1.58 when hepcidin is <10 ng/L) after admission to the intensive care unit.36 Hepcidin measurements were also evaluated in the differential diagnosis of anemia in inflammatory bowel disease patients, demonstrating that hepcidin <2 nmol/L identified iron deficiency better among these inflamed patients.37 Another study identified fasting morning serum hepcidin <2.4 nmol/L as a useful cutoff to distinguish between ACI in patients with concurrent iron deficiency relative to those without iron deficiency (area under the curve, 0.9; sensitivity 89%; specificity 88%).38 In contrast, studies using absent bone marrow iron to diagnose iron deficiency did not find hepcidin (measured by either mass spectrometry39 or ELISA40 ) to be more predictive than ferritin in elderly anemic patients. To enable a more universal application of hepcidin in the clinical setting requires the standardization of assays that have yet to be finalized.41
Several manuscripts provide evidence of hepcidin as a predictor of poor response to oral iron therapy. Because hepcidin negatively regulates iron absorption, suboptimally suppressed hepcidin has the potential to predict poor response to oral iron supplementation. Because patients on oral iron supplements with only minimal iron absorption are expected to have unabsorbed iron passing through the length of the intestine and unabsorbed iron may adversely impact the gut microbiome,42 hepcidin may be helpful both in avoiding this potentially adverse effect of unabsorbed iron supplementation and delaying initiation of parenteral iron. In iron deficiency anemia, hepcidin outperforms ferritin and transferrin saturation in predicting response to oral iron supplementation.43 Another study assessed hepcidin in elderly patients undergoing oral iron absorption testing, demonstrating a negative correlation between baseline hepcidin and the relative increase in transferrin saturation 4 hours after 160 mg oral iron and predicting effective absorption of oral iron in elderly patients with and without anemia and/or iron deficiency.38 Additional studies are underway to further elucidate the utility of hepcidin in various setting, including in chronic renal failure and dialysis patients, blood donors, and iron-deficient children in countries with a high burden of infectious diseases.
A recent review identified the most promising applications for hepcidin measurements: evaluation of patients with iron overload, suspected IRIDA, concomitant iron deficiency and ACI, and prediction of response to oral iron therapy.44 Because in iron deficiency anemia, hepcidin production is very low to maximize iron absorption,45 hepcidin concentrations in or above the normal range in the presence of iron deficiency would be considered a significant predictor of IRIDA, associated with a very low MCV anemia owing to iron-restricted erythropoiesis, and would confirm the requirement for parenteral iron to replete iron stores. Hepcidin may be suboptimally suppressed in the presence of iron deficiency anemia in additional conditions, especially in elderly patients30 with comorbid conditions (eg, renal, neoplastic, and inflammatory diseases). In concurrent iron deficiency anemia with ACI, hepcidin has also been found to provide individualized decision making regarding route of iron supplementation in anemia associated with rheumatoid arthritis,46 inflammatory bowel diseases,37 critical illness,47 or cancer48 and after chemotherapy.49
Available novel measurement options
The College of American Pathologists–approved and Clinical Laboratory Improvement Amendments–certified serum ELISA-based testing is available for measuring human hepcidin concentration.45 Other hepcidin assays using mass spectrometry platforms have also been developed.50 Although a number of hepcidin assays have been validated and correlate with one another, they show considerably different absolute hepcidin concentrations depending on the method of measurement. In light of this, significant work has been done to identify and resolve inconsistencies between the different assays, and a recent standardization demonstrates a potential for establishing an international hepcidin reference range.41 This standardization paves the way to implementing hepcidin into clinical practice. Thus, at this point in time, hepcidin measurements are available and can be used for clinical decision making. However, they are not reimbursed by insurance companies, requiring out-of-pocket contribution.
Taken together, although there is significant promise and several compelling evaluations already completed, the use of hepcidin to assist with clinical decision making in anemia evaluation remains limited to the research setting, and reasonable near-future goals in evaluating the utility of hepcidin measurements include determining meaningful validated reference ranges and cutoffs to assist clinicians in prescribed settings. In addition, because hepcidin regulation is a consequence of influences from systemic and cellular iron concentration, inflammation, hypoxia, and erythropoiesis, assessing the meaning of hepcidin measurement is likely to require context, in concert with several additional markers. Thus, the interpretation of hepcidin results requires further study, because there is no clearly defined meaningful cutoffs in all clinical scenarios to assist clinicians in complex cases, the very situations in which hepcidin measurements would be requested.
Case discussion
The patient has symptoms that could be attributed to either iron deficiency or chronic inflammation. Diagnosis of concurrent iron deficiency and ACI is also possible with increased menstrual bleeding resulting in iron deficiency and her recent pneumonia with associated inflammation causing ACI. The timing of anemia would also be consistent with both etiologies. Hospital records may assist in further evaluating the degree of contribution of inflammation to the anemia in this patient. Her ferritin does not meet criteria for systemic iron deficiency but her transferrin saturation suggests that she may have iron-restricted erythropoiesis. The fact that she does not have microcytic anemia does not rule out the possibility of iron deficiency, and the diagnostic approach would benefit from measuring sTfR1, CRP, ZPP/H, and HYPO% or CHr. Her sTfR1/log ferritin is >2, suggesting concurrent iron deficiency and ACI (Figure 5). The decision regarding whether to treat this patient with oral or parenteral iron is currently being decided based on patient preference/tolerability of oral iron supplementation and physician comfort with administration of parenteral iron. Based on evidence from multiple sources, hepcidin, if low (eg, <2 nmol/L or <20 ng/L), may be both useful to identify the presence of iron deficiency anemia even in inflammatory conditions and enable prediction of responsiveness to oral iron supplementation.28,37,47 Oral iron supplementation can be most gently accomplished with carbonyl iron caplets (45 mg each) as a single dose of 90 to 135 mg given every other day to maximize absorption.51 At this time, the best approach would be to give this patient a trial of oral iron supplementation for 3 to 6 months if well tolerated to enable good compliance. Alternatively, several parenteral iron formulations are available to enable rapid repletion of iron stores; this topic has recently been reviewed elsewhere.52
Unresolved questions and future directions
It is of relevance that there is no consensus even on serum ferritin cutoffs to define iron deficiency in some populations (eg, children and pregnant women). In light of this, all other metrics discussed herein fall significantly short, and the amalgam analysis of multiple markers remains to be systematically performed to demonstrate predictability, reliability in therapeutic decision making, and relevance to health outcomes. For example, the role of sTfR1 in the physiology of iron deficiency and iron-restricted erythropoiesis is not clearly understood. Although it is used in differentiating ACI from iron deficiency anemia with chronic inflammation, increased sTfR1 may occur either when erythropoiesis is expanded (increased number of erythroblasts with unchanged TfR1 expression per cell) or in iron deficiency erythropoiesis (unchanged number of erythroblasts but increased TfR1 expression per cell). Thus, understanding the function of sTfR1 in iron metabolism would greatly benefit a refined method of use in diagnosis and possibly treatment of anemias in general and ACI in particular.
Additional research is also needed to evaluate how ERFE can contribute to our understanding, diagnosis, and management of iron deficiency and iron-restricted erythropoiesis in the context of ACI. A dual monoclonal antibody sandwich ELISA is also available for human serum ERFE53 but has not yet received standard approval and certification by the regulatory agencies required for clinical use. Recent work demonstrates superiority of ERFE in predicting acute decompensated heart failure and heart failure–related death relative to more standard metrics,54 and elevated ERFE correlates with cardiovascular events in hemodialysis and prehemodialysis renal disease patients.55 Recent literature reveals that iron deficiency is observed in a high fraction of heart failure patients,56 and parenteral iron reduces hospitalization57 and mortality rates in these patients.58 It is thus tempting to speculate that an element of ineffective erythropoiesis, resulting in increased ERFE, may be the cause of anemia or consequence of systemic inflammation during heart failure exacerbation. Whether and how ERFE functions as a specific biomarker in acute heart failure or other nonhematologic diseases remain to be determined, and whether its elevation defines a subpopulation that would specifically benefit from iron supplementation is unknown.
Lastly, the pace of translating hepcidin from a newly identified regulator of iron metabolism to its manipulation for diagnostic and therapeutic purposes within the span of a decade is tremendous progress for the entire field. Significantly more work still remains to be done to enable the rational use of hepcidin to supplement the currently available diagnostic tools in a meaningful way. In addition to its promising role in supporting the diagnosis of several iron-related diseases,44 studies are underway to determine whether hepcidin is a useful marker to predict response to oral iron supplementation in iron-deficient patients59 and response to erythropoiesis stimulating agents (eg, aranesp, procrit) in hemodialysis patients.60 Its anticipated utility would enable (1) more definitive diagnosis of iron deficiency in the context of inflammation, (2) further refined patient selection for parenteral iron supplementation (if it can predict poor response to oral iron supplementation in iron-deficient patients), and (3) enhanced and perhaps individualized dosing schema for and duration of iron supplementation.
Acknowledgments
The author thanks Laura Silvestri, PhD, for assistance with the figures; advocates and mentors for support in the past, present, and future; and the patients who lay their trust in us.
Correspondence
Yelena Z. Ginzburg, Tisch Cancer Institute, Division of Hematology and Medical Oncology, Icahn School of Medicine at Mount Sinai, 1 Gustave L. Levy Place, Box 1079, New York, NY 10029; e-mail: yelena.ginzburg@mssm.edu.
