
Abstract
The treatment of primary immunodeficiency disorders with allogeneic hematopoietic cell transplantation (HCT) has a history dating back to 1968 with the first successful transplant for a patient with severe combined immunodeficiency (SCID). The omission of conditioning for patients with SCID owing to their inability to reject allogeneic grafts and the increasing use of reduced intensity conditioning regimens often result in a state of mixed or split donor-recipient chimerism. The use of gene therapy (GT) via retroviral or lentiviral transduction of autologous CD34+ hematopoietic stem and progenitor cells is expected to correct only a portion of the hematopoietic stem cell compartment. The consequences of partial correction after either form of cellular therapy differ according to how the genetic deficiency affects immune cell development and function. Moreover, the conditioning regimen or lack thereof impacts the cell lineages at risk of partial correction. Advances in our understanding of immune reconstitution after HCT and GT for SCID, Wiskott–Aldrich syndrome, and chronic granulomatous disease are discussed.
Learning Objectives
Understand the principles and impact of conditioning used for patients with primary immunodeficiency on mixed chimerism posttransplant
Describe the role of selective advantage, conditioning, and genotype on outcome in patients with severe combined immunodeficiency
Explain the impact of mixed chimerism and mechanisms of autoimmunity posttreatment in patients with Wiskott–Aldrich syndrome
Understand that partial correction of neutrophils in chronic granulomatous disease after treatment may not control all symptoms
Clinical case
A 2-week-old male infant, the first child born to nonrelated parents, was called back after discharge from the newborn nursery because of newborn screen suspicious for severe combined immunodeficiency (SCID). Evaluation confirms the absence of T cells, normal numbers of B cells, and poor proliferation to mitogens, and later, it reveals him to have mutation in IL2RG. What are his options for treatment? What conditioning should be used before therapy? What is the optimal choice to restore full T- and B-cell numbers and function?
Introduction
Primary immunodeficiency disorders (PIDs) comprise a broad range of disorders affecting immune cell development and function, and increasingly, they are known to have monogenic causes. The International Union of Immunological Societies classifies the 354 distinct disorders into 9 categories,1 and those commonly treated with allogeneic hematopoietic cell transplantation (HCT) generally fall into categories of combined immunodeficiency, immunodeficiency with syndromic features, immune dysregulation, and phagocyte number or function (Table 1). With greater understanding of the molecular underpinnings of these complex disorders, we now have the opportunity to move away from a one-size-fits-all approach to cellular therapy. This includes tailoring of the conditioning regimen to take advantage of lineage-specific selective advantage, consideration of the individual gene defect within a category of similar diseases, and the use of autologous gene therapy (GT) for diseases of known genetic cause. Advances in the 5 to 10 years of cellular therapy for SCID, Wiskott–Aldrich syndrome (WAS), and chronic granulomatous disease (CGD) will be discussed.
Genetically characterized PID for which HCT is well established as treatment
| Disorder | Gene(s) |
|---|---|
| Cellular and humoral immunity | |
| SCID | Multiple (Table 3) |
| X-linked hyper-IgM syndrome | CD40LG |
| DOCK8 deficiency | DOCK8 |
| ZAP70 deficiency | ZAP70 |
| Combined immunodeficiency with associated or syndromic features | |
| Wiskott–Aldrich syndrome | WAS |
| Dyskeratosis congenita | DKC1, NHP2, NOP10, RTEL1, TERC, TERT, TINF2, ACD, PARN, WRAP53, STN1, CTC1, NAF1 |
| NEMO deficiency | IKBKG |
| Diseases of immune dysregulation | |
| Hemophagocytic lymphohistiocytosis | PRF1, UNC13D, STX11, STXBP2 |
| Other syndromes at risk for hemophagocytic lymphohistiocytosis (Chediak–Higashi syndrome, Griscelli syndrome type 2) | LYST, RAB27A |
| Immune dysregulation, polyendocrinopathy, enteropathy, X-linked | FOXP3 |
| X-linked lymphoproliferative disease | SH2D1A, XIAP |
| Defects of phagocytes | |
| Chronic granulomatous disease | CYBB, CYBA, NCF1, NCF2 |
| Severe congenital neutropenia | ELANE, GFI1, HAX1 |
| Leukocyte adhesion deficiency type 1 | ITGB2 |
| Defects of IL-10 pathway | IL10RA, IL10RB |
| MonoMac syndrome | GATA2 |
| Defects in intrinsic and innate immunity | |
| Mendelian susceptibility to mycobacterial disease | IFNGR1, IFNGR2 |
| Osteopetrosis | TCIRG1, TNFRSF11A (RANK) |
| Disorder | Gene(s) |
|---|---|
| Cellular and humoral immunity | |
| SCID | Multiple (Table 3) |
| X-linked hyper-IgM syndrome | CD40LG |
| DOCK8 deficiency | DOCK8 |
| ZAP70 deficiency | ZAP70 |
| Combined immunodeficiency with associated or syndromic features | |
| Wiskott–Aldrich syndrome | WAS |
| Dyskeratosis congenita | DKC1, NHP2, NOP10, RTEL1, TERC, TERT, TINF2, ACD, PARN, WRAP53, STN1, CTC1, NAF1 |
| NEMO deficiency | IKBKG |
| Diseases of immune dysregulation | |
| Hemophagocytic lymphohistiocytosis | PRF1, UNC13D, STX11, STXBP2 |
| Other syndromes at risk for hemophagocytic lymphohistiocytosis (Chediak–Higashi syndrome, Griscelli syndrome type 2) | LYST, RAB27A |
| Immune dysregulation, polyendocrinopathy, enteropathy, X-linked | FOXP3 |
| X-linked lymphoproliferative disease | SH2D1A, XIAP |
| Defects of phagocytes | |
| Chronic granulomatous disease | CYBB, CYBA, NCF1, NCF2 |
| Severe congenital neutropenia | ELANE, GFI1, HAX1 |
| Leukocyte adhesion deficiency type 1 | ITGB2 |
| Defects of IL-10 pathway | IL10RA, IL10RB |
| MonoMac syndrome | GATA2 |
| Defects in intrinsic and innate immunity | |
| Mendelian susceptibility to mycobacterial disease | IFNGR1, IFNGR2 |
| Osteopetrosis | TCIRG1, TNFRSF11A (RANK) |
IgM, immunoglobulin M; IL-10, interleukin-10; NEMO, NF-κB essential modulator.
Principles of conditioning for PID patients
Unlike patients with malignancy undergoing HCT, where a major goal of conditioning is to eliminate residual malignancy, the major purposes of conditioning for patients with PID undergoing HCT are to eliminate recipient T cells and natural killer (NK) cells, thereby preventing rejection of donor-derived hematopoietic stem cells (HSCs), and at least partially myeloablate the patient, providing sufficient space in the recipient bone marrow for those stem cells to engraft to a level that is curative for the patient’s particular disease. A regimen of busulfan for myeloablation combined with cyclophosphamide for T-cell ablation reported to be successful for full immune reconstitution in 3 patients with WAS became the basic backbone for the vast majority of conditioning regimens for nonmalignant disease.2
SCID is an exception to the rule that conditioning is always required for allogeneic HCT as demonstrated >50 years ago.3 Patients with typical SCID regardless of the genetic cause have a profound absence of T cells owing to failure of T-cell development in the thymus, and as such, SCID patients generally lack the ability to reject an allogeneic graft, particularly when a fully HLA-matched sibling donor is used. Furthermore, HSCs from the donor can seed the thymus (which in many forms of SCID, lacks T-cell progenitors), engraft in the “empty” niche, and generate donor-derived T cells for many decades, despite the lack of engraftment of donor-derived HSCs in the bone marrow (Figure 1). The inability of SCID patients to reject allogeneic cells is so profound that maternal-fetal blood exchange in utero can result in maternal T-cell engraftment. Haploidentical bone marrow from a parent can also be infused without conditioning and successfully reconstitute T-cell development as long as the graft is vigorously T cell depleted to prevent graft-versus-host disease (GVHD).4,5 T-cell reconstitution after haploidentical transplant is less reliable than after matched sibling donor transplant, at least in part because of the capacity of NK cells to reject mismatched grafts.

The effect of SCID subtype and use of conditioning on T- and B-cell chimerism. Patients with SCID who undergo HCT without conditioning have engraftment of donor-derived cells in the thymus, giving rise to donor-derived T cells (purple). HSCs and B cells, if present, remain largely of recipient origin (blue). With conditioning, donor HSCs can engraft, giving rise to donor-derived B cells, and also, they can continuously replenish progenitors in the thymus, resulting in robust and durable T-cell production.
The effect of SCID subtype and use of conditioning on T- and B-cell chimerism. Patients with SCID who undergo HCT without conditioning have engraftment of donor-derived cells in the thymus, giving rise to donor-derived T cells (purple). HSCs and B cells, if present, remain largely of recipient origin (blue). With conditioning, donor HSCs can engraft, giving rise to donor-derived B cells, and also, they can continuously replenish progenitors in the thymus, resulting in robust and durable T-cell production.
Mechanisms of mixed T-cell chimerism after HCT
The use of high-dose regimens, such as busulfan and cyclophosphamide, in infants and children with PID has long-term impact on linear growth, fertility, and secondary malignancy. The use of individualized busulfan pharmacokinetics to target different exposures to drug, the substitution of cyclophosphamide with fludarabine, and the use of lympholytic antibodies, such as antithymocyte globulin (ATG) or alemtuzumab, have led to the development of reduced toxicity regimens. In general, conditioning regimens with a reduced dose of myeloablative agents carry a risk of mixed chimerism in the HSC compartment and therefore, mixed chimerism in other compartments, such as neutrophils, B cells, red blood cells, and platelets.
The impact of conditioning regimen on T-cell reconstitution is more complex. T cells reconstitute after allogeneic transplant by extrathymic and thymic pathways (Figure 2A). After a T cell–replete HCT, mature donor–derived T cells infused with the graft expand extrathymically and are potent drivers of GVHD. In contrast to infused T cells, donor-derived T cells can also mature through a thymic pathway. Donor-derived HSCs seed the recipient’s thymus and are induced to commit to the T-cell lineage by interaction with thymic epithelium. The T cells that then are exported from the thymus have a naïve nonactivated phenotype, are restricted to the recipient’s HLA, and are tolerant to the host. Development of T cells through the thymic pathway generally is not detectable until 4 to 6 months post-HCT, and it is much delayed in adults compared with children owing to involution and decreased activity of the thymus with age.6
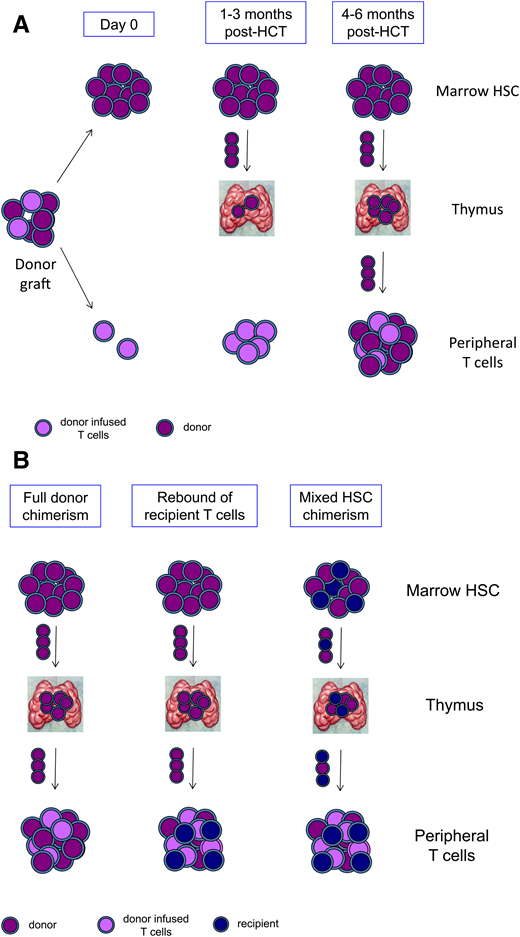
T-cell reconstitution and mixed chimerism after HCT. (A) The donor graft contains HSCs and progenitors that engraft in the marrow (purple) and mature T cells that have been tolerized to the donor and are adoptively transferred (lavender). Over the first 1 to 3 months post-HCT, transferred mature T cells proliferate extrathymically, and donor HSCs seed the thymus. Beginning at 4 to 6 months post-HCT, newly generated donor-derived naïve T cells emerge from the thymus and populate the periphery. (B) In states of full donor chimerism (left panel), marrow HSCs, thymocytes, and peripheral T cells are all donor derived (purple). Rebound of recipient T cells (blue) may occur despite full donor chimerism in HSCs and the development of donor T cells from the thymus (center panel). Mixed chimerism in T cells may also arise if there is mixed chimerism in HSCs, resulting in mixed chimerism in thymocytes and in naïve T cells emerging from the thymus (right panel).
T-cell reconstitution and mixed chimerism after HCT. (A) The donor graft contains HSCs and progenitors that engraft in the marrow (purple) and mature T cells that have been tolerized to the donor and are adoptively transferred (lavender). Over the first 1 to 3 months post-HCT, transferred mature T cells proliferate extrathymically, and donor HSCs seed the thymus. Beginning at 4 to 6 months post-HCT, newly generated donor-derived naïve T cells emerge from the thymus and populate the periphery. (B) In states of full donor chimerism (left panel), marrow HSCs, thymocytes, and peripheral T cells are all donor derived (purple). Rebound of recipient T cells (blue) may occur despite full donor chimerism in HSCs and the development of donor T cells from the thymus (center panel). Mixed chimerism in T cells may also arise if there is mixed chimerism in HSCs, resulting in mixed chimerism in thymocytes and in naïve T cells emerging from the thymus (right panel).
Mixed chimerism in the T-cell compartment can occur through different mechanisms (Figure 2B). Recipient T cells that fail to be eliminated by conditioning agents targeting T cells (fludarabine, cyclophosphamide, irradiation, ATG, and alemtuzumab) may rebound and expand post-HCT. Recent studies of pharmacokinetics of fludarabine, ATG, and alemtuzumab, including modeling specifically in small children, have shown the negative impact of over- or underdepletion of lymphocytes.7-10 For example, residual free ATG remaining at day 0 has been shown to be strongly associated with engraftment, rapid T-cell reconstitution, and survival, and predictors of ATG metabolism and T-cell reconstitution in individual patients are increasingly understood.11
Mixed T-cell chimerism may also arise because of mixed chimerism at the HSC level, leading to intrathymic development of both donor-derived and recipient-derived T cells (Figure 2B). Because de novo generation of T cells takes many months, such mixed T-cell chimerism would only be evident late post-HCT at 6 to 12 months or later. The contribution of long-term HSCs to mature cell output can only be assessed at 6 to 12 months post-HCT or later after output from shorter-lived progenitors from the donor has dissipated. For this reason, HSC chimerism in the first year post-HCT is unstable. Mixed HSC chimerism at late time points will result in mixed T-cell chimerism, though only in situations where neither recipient- nor donor-derived T cells have a selective survival or developmental advantage.
Autologous GT as an alternative to HCT
The delineation of the monogenetic causes for PID, development of retroviral and lentiviral integrating vectors, and technological advances in isolation and transduction of human HSCs have fueled the growth of autologous GT as a viable alternative to HCT for the treatment of PID. Table 2 lists PIDs for which efficacious GT trial results have been published and the status of regulatory approvals for licensure.
Completed, active in follow-up, and recruiting GT trials in SCID, WAS, and CGD
| Promoter/transgene | NCT no. | Status | Vector type | Ref. | Center(s) | Nonacademic sponsorship or commercialization |
|---|---|---|---|---|---|---|
| ADA-deficient SCID | ||||||
| γRV-ADA | NCT00599781 | Completed | γRV | 15,16 | OSR-TIGET | Marketed as Strimvelis in Europe |
| γRV-ADA | NCT01279720 | Completed | γRV | 17 | GOSH | |
| MND-γRV-ADA | NCT00794508 | Completed | γRV | 18,19 | UCLA | |
| GCsapM-γRV-ADA | NCT00018018 | Completed | γRV | NHGRI | ||
| EFS-codon optimized ADA | NCT01852071 | Completed | LV | 31 | UCLA, NHGRI | Sponsored by Orchard Therapeutics, OTL-101 |
| NCT02999984 | Active, not recruiting | LV | UCL, UCLA | |||
| NCT01380990 | Active, not recruiting | LV | GOSH | |||
| NCT03765632 | Recruiting | LV | GOSH | |||
| X-linked SCID | ||||||
| EFS-IL2RG | NCT01129544 | Active, not recruiting | SIN-γRV | 14 | BCH, UCLA, CCHMC | |
| NCT01410019 | Unknown | HNEM | ||||
| NCT01175239 | Unknown | GOSH | ||||
| EFS codon-optimized IL2RG | NCT03311503 | Recruiting | LV | BCH, UCLA | ||
| NCT03601286 | Recruiting | LV | GOSH | |||
| EFS codon-optimized IL2RG with insulator | NCT01306019 | Recruiting | LV | 36 | NIAID | |
| NCT01512888 | Recruiting | LV | 37 | SJCRH | ||
| UCSF | ||||||
| SCRI | ||||||
| EFS codon-optimized IL2RG with insulator | NCT03315078 | Recruiting | LV | NIAID | ||
| IL2RG, not specified | NCT03217617 | Recruiting | LV | SG-IMI | ||
| WAS | ||||||
| 1.6-kb WAS promoter-hWAS | NCT01515462 | Active, not recruiting | LV | 40 | OSR-TIGET | Sponsored by Orchard Therapeutics, OTL-103 |
| NCT03837483 | Recruiting | LV | OSR-TIGET | Sponsored by Orchard Therapeutics, OTL-103 | ||
| NCT01347346 | Completed | LV | 41 | HNEM | Sponsored by Genethon | |
| NCT01347242 | Recruiting | LV | GOSH | Sponsored by Genethon | ||
| NCT01410825 | Active, not recruiting | LV | BCH | |||
| X-linked CGD | ||||||
| γRV-CYBB | NCT00927134 | Completed | γRV | 53,54 | UCHZ | |
| Cathepsin G/c-fes chimeric promoter codon-optimized CYBB | NCT02234934 | Recruiting | LV | UCLA, BCH, NIAID | ||
| NCT02757911 | Recruiting | LV | HNEM | Sponsored by Genethon | ||
| NCT01855685 | Recruiting | LV | UCL, RFH, GOSH | Sponsored by Genethon | ||
| CYBB, not specified | NCT03645486 | Recruiting | LV | SG-IMI | ||
| CYBB, not specified | NCT01906541 | Recruiting | SIN-γRV | UHF | ||
| CYBB, not specified | NCT00778882 | Active, not recruiting | γRV | SNU | Sponsored by Helixmith |
| Promoter/transgene | NCT no. | Status | Vector type | Ref. | Center(s) | Nonacademic sponsorship or commercialization |
|---|---|---|---|---|---|---|
| ADA-deficient SCID | ||||||
| γRV-ADA | NCT00599781 | Completed | γRV | 15,16 | OSR-TIGET | Marketed as Strimvelis in Europe |
| γRV-ADA | NCT01279720 | Completed | γRV | 17 | GOSH | |
| MND-γRV-ADA | NCT00794508 | Completed | γRV | 18,19 | UCLA | |
| GCsapM-γRV-ADA | NCT00018018 | Completed | γRV | NHGRI | ||
| EFS-codon optimized ADA | NCT01852071 | Completed | LV | 31 | UCLA, NHGRI | Sponsored by Orchard Therapeutics, OTL-101 |
| NCT02999984 | Active, not recruiting | LV | UCL, UCLA | |||
| NCT01380990 | Active, not recruiting | LV | GOSH | |||
| NCT03765632 | Recruiting | LV | GOSH | |||
| X-linked SCID | ||||||
| EFS-IL2RG | NCT01129544 | Active, not recruiting | SIN-γRV | 14 | BCH, UCLA, CCHMC | |
| NCT01410019 | Unknown | HNEM | ||||
| NCT01175239 | Unknown | GOSH | ||||
| EFS codon-optimized IL2RG | NCT03311503 | Recruiting | LV | BCH, UCLA | ||
| NCT03601286 | Recruiting | LV | GOSH | |||
| EFS codon-optimized IL2RG with insulator | NCT01306019 | Recruiting | LV | 36 | NIAID | |
| NCT01512888 | Recruiting | LV | 37 | SJCRH | ||
| UCSF | ||||||
| SCRI | ||||||
| EFS codon-optimized IL2RG with insulator | NCT03315078 | Recruiting | LV | NIAID | ||
| IL2RG, not specified | NCT03217617 | Recruiting | LV | SG-IMI | ||
| WAS | ||||||
| 1.6-kb WAS promoter-hWAS | NCT01515462 | Active, not recruiting | LV | 40 | OSR-TIGET | Sponsored by Orchard Therapeutics, OTL-103 |
| NCT03837483 | Recruiting | LV | OSR-TIGET | Sponsored by Orchard Therapeutics, OTL-103 | ||
| NCT01347346 | Completed | LV | 41 | HNEM | Sponsored by Genethon | |
| NCT01347242 | Recruiting | LV | GOSH | Sponsored by Genethon | ||
| NCT01410825 | Active, not recruiting | LV | BCH | |||
| X-linked CGD | ||||||
| γRV-CYBB | NCT00927134 | Completed | γRV | 53,54 | UCHZ | |
| Cathepsin G/c-fes chimeric promoter codon-optimized CYBB | NCT02234934 | Recruiting | LV | UCLA, BCH, NIAID | ||
| NCT02757911 | Recruiting | LV | HNEM | Sponsored by Genethon | ||
| NCT01855685 | Recruiting | LV | UCL, RFH, GOSH | Sponsored by Genethon | ||
| CYBB, not specified | NCT03645486 | Recruiting | LV | SG-IMI | ||
| CYBB, not specified | NCT01906541 | Recruiting | SIN-γRV | UHF | ||
| CYBB, not specified | NCT00778882 | Active, not recruiting | γRV | SNU | Sponsored by Helixmith |
BCH, Boston Children’s Hospital, Boston, MA; CCHMC, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH; EFS, elongation factor 1a short; γRV, gammaretrovirus; GOSH, Great Ormond Street Hospital, London, United Kingdom; HNEM, Hopital Necker Enfants Malades, Paris, France; LV, lentivirus; NHGRI, National Human Genome Research Institute, Bethesda, MD; NIAID, National Institute of Allergy and Infectious Diseases, Bethesda, MD; OSR-TIGET, Ospedale San Raffaele–Telethon Institute for Gene Therapy, Milan, Italy; RFH, Royal Free Hospital, London, United Kingdom; SCRI, Seattle Children’s Research Institute, Seattle, WA, USA; SG-IMI, Shenzhen Geno-Immune Medical Institute, Shenzhen, People’s Republic of China; SIN, self-inactivating; SJCRH, St. Jude Childhood Research Institute, Memphis, TN; SNU, Seoul National University, Seoul, Republic of Korea; UCHZ, University Children’s Hospital Zurich, Zurich, Switzerland; UCL, University College London, London, United Kingdom; UCLA, University of California, Los Angeles, CA; UCSF, University of California, San Francisco, CA; UHF, University Hospital Frankfurt, Frankfurt, Germany.
The safety of GT has greatly improved in recent years. Early trials using unmodified gammaretroviral vectors containing intact virus-derived promoters and enhancers were associated with insertional oncogenesis and the development of lymphoid and myeloid malignant transformation owing to integration near and transactivation of proto-oncogenes, such as LMO2 and MECOM.12,13 A multi-institutional trial of GT for X-linked SCID using an enhancer-deleted self-inactivating gammaretroviral vector reported efficacy without insertional oncogenesis,14 which is sustained with longer follow-up (S.-Y. Pai, unpublished data, 6 August 2019). Interestingly, trials using unmodified gammaretroviral vectors in ADA SCID have never been associated with insertional oncogenesis, despite abundant integrations near LMO2 and MECOM.15-20 Nevertheless, the vast majority of trials of GT for PID using HSCs have since used self-inactivating lentiviral vectors, with no insertional oncogenesis events reported to date, likely because of the distinct and more neutral insertion pattern of lentiviruses compared with gammaretroviruses.
Autologous GT obviates the need to identify a well-matched donor and eliminates the risk of GVHD. The conditioning regimen can also be simplified in that agents to prevent allogeneic rejection may be omitted. Ensuring that GT is equally efficacious as HCT remains challenging because of factors intrinsic to the GT approach. High-dose chemotherapy regimens (eg, busulfan with pharmacokinetic targeting to myeloablative levels) are unlikely to eliminate all recipient HSCs in every patient. Allogeneic graft-vs-marrow effect that promotes engraftment after HCT does not contribute in the autologous GT setting. Transduction efficiency, measured as the percentage of colony-forming units of cultured cells from the medicinal product, is typically 30% to 90%, not 100%, and therefore, autologous GT remains likely to result in incomplete correction of HSCs. Within transduced cells, copy number may vary from 1 to 5 copies per cell or more, resulting in variable expression of the transgene. Depending on the promoter used to drive transgene expression (viral, ubiquitous, cell-type specific, or endogenous to the transgene), the level of expression of the transgene in a cell carrying 1 copy of the vector may be lower than wild-type expression. Conversely, some vectors in clinical trial have been engineered to be codon optimized to increase the efficiency of translation and yield higher transgene protein levels per copy of gene inserted (Table 2).
Outcomes of HCT and GT for SCID
The inability of patients with typical SCID to reject a matched sibling donor allogeneic graft combined with the natural ability of HSCs and other progenitors to enter the thymus and develop into T cells underlies the success of HCT in SCID patients in the absence of conditioning. Such transplants generally result in a state of what is properly termed split chimerism in that the T cells will be solely donor derived, because the recipient lacks the capacity to generate T cells, whereas the HSCs and all other progeny of HSCs are usually recipient derived (Figure 1). As long as mature T cells arise and are sustained after HCT, the patient is rescued from life-threatening opportunistic infection.
Factors related to survival and immune reconstitution of patients with SCID after HCT are increasingly understood owing to registry studies in Europe (Stem Cell Transplantation for Immunodeficiencies [SCETIDE] registry) and North America (Primary Immune Deficiency Treatment Consortium [PIDTC]).21-24 Overall survival after HCT is 70% or greater in the most modern cohorts, and matched sibling donor recipients consistently fare the best, with >90% survival. Survival after HCT for SCID when using alternative donors (T cell–depleted haploidentical donor, adult volunteer unrelated donor, or unrelated umbilical cord blood) remains equivalent.21-24 Although no one donor source has emerged as superior, unrelated umbilical cord blood transplant recipients demonstrate a trend to lower survival in multiple studies.21-24 Other critical factors driving survival include age at transplant, with those transplanted at age <3.5 months old having superior survival, and the presence of active infection at the time of HCT, which was associated with poor survival.22,24,25
The genetic causes of SCID can be divided broadly into those defects that are intrinsic to HSC vs those that are caused by defects in thymic epithelium (Table 3). In North America, where all 50 states in the United States and some provinces in Canada are screening for SCID at birth by assaying T-cell receptor excision circles in dried blood spots, a genetic cause can be identified in >90% of cases of SCID undergoing HCT.26 Understanding the nature of the block in T-cell development and the impact on B-cell development and function in specific genotypes informs the potential impact of split chimerism on immune function post-HCT and in turn, which patients may require conditioning for full immune reconstitution. In a recent PIDTC study of 662 patients with SCID, of whom 571 had undergone HCT with a donor other than a matched sibling, patients with defects in ADA or DCLRE1C and those with unknown defects had poorer survival compared with patients with X-linked SCID (caused by defects in IL2RG) or defects in JAK3 (the signaling component downstream of IL2RG).22 Different genotypes were at risk of poor T-cell reconstitution than were at risk of poor B-cell function post-HCT, and both T- and B-cell reconstitution was positively associated with the use of conditioning.22-24 Patients with defects in recombination of T- and B-cell receptors (RAG1, RAG2, or DCLRE1C), which result in the absence of T cells owing to arrest in development in CD4− CD8− thymocytes,27 had poorer T-cell numbers and/or function at 2 to 5 years post-HCT than other genotypes. This may be owing to NK cell–mediated rejection, particularly of mismatched grafts, in the setting of aberrant and overactive NK activity in RAG-mutated patients.28 The presence of RAG- or DCLRE1C-deficient early thymocytes may interfere with engraftment of HSC and progenitors owing to occupancy of the thymic epithelial “niche.” Conditioning to overcome these barriers to T-cell reconstitution thus must be successful at eliminating host NK cells and host thymocytes.
Genetic causes of SCID
| HSC intrinsic defects | Thymic epithelium defects | |||
|---|---|---|---|---|
| B cells present (B+ SCID) | B cells absent (B− SCID) | |||
| B cells defective | B cells functional | Not radiosensitive | Radiosensitive | |
| IL2RG (X-linked) JAK3 | IL7R | RAG1 | DCLRE1C (Artemis) | DiGeorge syndrome (22q11 deletion and others) |
| CD3D | RAG2 | LIG4 | CHD7 (CHARGE syndrome) | |
| CD3E | ADA | NHEJ1 (Cernunnos) | FOXN1 | |
| PTPRC (CD45) | AK2 (reticular dysgenesis) | NBS1 (Nijmegen breakage syndrome) | ||
| HSC intrinsic defects | Thymic epithelium defects | |||
|---|---|---|---|---|
| B cells present (B+ SCID) | B cells absent (B− SCID) | |||
| B cells defective | B cells functional | Not radiosensitive | Radiosensitive | |
| IL2RG (X-linked) JAK3 | IL7R | RAG1 | DCLRE1C (Artemis) | DiGeorge syndrome (22q11 deletion and others) |
| CD3D | RAG2 | LIG4 | CHD7 (CHARGE syndrome) | |
| CD3E | ADA | NHEJ1 (Cernunnos) | FOXN1 | |
| PTPRC (CD45) | AK2 (reticular dysgenesis) | NBS1 (Nijmegen breakage syndrome) | ||
CHARGE, coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, ear abnormalities.
The effect of genotype on B-cell function post-HCT can be explained by differential requirements for donor-derived B-cell development (Figure 1). In general, B-negative forms of SCID require conditioning to have successful humoral immune function. Unlike T cells, the production of which can be sustained over decades by progenitors engrafting in the thymus, sustained production of B cells requires engraftment of long-term HSCs. Patients with genetic defects that abrogate B-cell receptor rearrangement in particular (RAG1, RAG2, DCLRE1C, LIG4, NHEJ1, and NBS1) were less likely to be independent of immunoglobulin substitution at 2 to 5 years post-HCT in PIDTC studies.22,24 For defects that also are associated with radiosensitivity and reduced capacity for DNA repair after exposure to alkylating agents, such as busulfan, high-dose conditioning is often avoided, which in turn, may compromise immune reconstitution.29,30
Patients with ADA deficiency also have low B-cell numbers owing instead to poor survival and cytotoxicity caused by accumulation of 2′-deoxyadenoside triphosphate. Restoration of B-cell function in ADA patients was more likely to occur than in patients with B-negative SCID owing to recombination defects at 2 to 5 years post-HCT.22 Several factors may contribute to the relative ease of correction of immune function in ADA patients compared with other forms of B-negative SCID. Donor-derived lymphocytes can crosscorrect ADA-deficient lymphocytes by detoxifying metabolites in the environment. Unlike recombination-defective B precursors, which accumulate at the pro–B-cell stage and then, suffer an arrest, the survival of ADA-deficient B-cell progenitors is affected all along the developmental pathway, providing selective advantage at every stage. The HSCs of ADA-deficient patients may themselves have a selective disadvantage, promoting engraftment of donor-derived HSCs even with low-dose conditioning.
The relative ease of effecting immune reconstitution in patients with ADA-SCID post-HCT contributes to the dramatic progress of the exposure needed for myeloablation in GT for this disease. Low-dose busulfan conditioning with ≤25% exposure than that needed for myeloablation resulted in engraftment of gene-marked cells in multiple trials.15-19 Furthermore, the use of conditioning and enhanced engraftment of marked HSCs leads to a diversification of the insertion site pattern.20 Codon optimization of the ADA transgene and expression of supraphysiologic levels of enzyme also improved efficacy in the face of low-level copy number integration.31 Gammaretroviral GT for ADA deficiency was commercialized as Strimvelis in 2016 in Europe, and lentiviral GT is on the path to commercialization in the United States (Table 2).
In contrast, defects in cytokine receptors (IL2RG, JAK3, and IL7R), components of the CD3 complex (CD3D and CD3E), and CD45 (PTPRC) preserve B-cell development. Yet, patients with IL2RG/JAK3 SCID are less likely to be independent of immunoglobulin at 2 to 5 years compared with IL7R/CD3/CD45-deficient patients.22 Although these latter defects should have no effect on B-cell function, defects in IL2RG/JAK3 impair the ability of B cells to respond to interleukin-21 (IL-21), a cytokine critical for B-cell maturation, class switching, and secretion of immunoglobulin signaling. Cells from patients with IL2RG or JAK3 SCID who have not received conditioning or who lack donor-derived B cells fail to differentiate or secrete immunoglobulin in vitro in response to IL-21, and in turn, those patients remain dependent on immunoglobulin replacement.32 Conditioning is thus required to promote engraftment of donor-derived HSCs and generation of donor-derived B cells to restore IL-21 response. The minimum level of busulfan needed to reliably result in T- and B-cell reconstitution in patients with IL2RG/JAK3 and RAG1/RAG2 SCID is being tested in a PIDTC randomized clinical trial of low vs moderate busulfan conditioning (NCT03619551).
This principle is further supported by the experience of GT for X-linked SCID. T cells carrying the IL2RG transgene consistently develop in the absence of conditioning; however, little to no gene marking in seen in B cells, rendering patients largely dependent on immunoglobulin replacement. In contrast, those who have received low-dose busulfan conditioning, including older adult patients with poor immune reconstitution posthaploidentical transplant, have B-cell marking and evidence of B-cell function.14,33-36 Currently, multiple open trials for X-linked SCID are testing lentiviral vectors with codon-optimized transgenes with low-dose busulfan (Table 2).
Outcomes of HCT and GT for WAS
WAS, an X-linked disorder caused by mutations in the WAS gene, results in defects of actin cytoskeleton rearrangement in T cells, B cells, NK cells, monocytes, dendritic cells, and platelets and deficiency, manifesting clinically as immunodeficiency, eczema, microthrombocytopenia, autoimmunity, and lymphoma. Multiple lines of evidence demonstrate that Wiskott–Aldrich syndrome protein (WASP) expression confers a strong selective advantage to T cells, NK cells, and certain subpopulations of B cells but little to no advantage to myeloid cells.37
Historical reports of HCT for WAS demonstrated the critical requirement for both immunoablation and myeloablation, with early experiences showing reconstitution of immune cells but failure of platelet reconstitution.37 The same strong selective advantage for lymphocytes, particularly T lymphocytes, underlies the relative ease with which this compartment and therefore, the symptoms of immunodeficiency and eczema are corrected compared with thrombocytopenia. In a report of 194 patients with WAS reviewed retrospectively, donor chimerism was higher in T cells than in B cells or myeloid cells.38 Mixed chimerism in myeloid cells as a surrogate for mixed chimerism in the HSC compartment was associated with thrombocytopenia, particularly when chimerism was below 50%.38 The strong selective advantage for T cells expressing WAS is also apparent after autologous GT, where T-cell marking is consistently higher in individual patients than in myeloid cells.39,40
Autoimmune manifestations in WAS are broad, including autoimmune hemolytic anemia, autoimmune neutropenia, immune thrombocytopenia, inflammatory bowel disease, vasculitis of the skin, arthritis, and immunoglobulin A nephropathy. The pathophysiology of autoimmunity in WAS is complex in that deficiencies in cell number or cell function in multiple lineages contribute. Regulatory T cells (Tregs) are present in normal numbers in WAS patients, but they are defective in their ability to suppress effector T cells in both humans and mouse models.37 Similar to other T cells, WASP-deficient Tregs have a survival disadvantage and are outcompeted by WASP-expressing Tregs in WAS carriers and WAS-deficient mice.37 WASP-deficient B cells are dysregulated, form spontaneous germinal centers, and produce a broad range of autoantibodies.41-43 The B-cell intrinsic nature of this dysregulation was supported by studies in mice rendered deficient only in B cells that develop autoimmune disease in the face of normal T cells and Tregs.41,43 Two groups have reported that regulatory B cells (Bregs), which produce the immunoregulatory cytokine IL-10, were substantially reduced in WAS patients and WAS-deficient mice.44,45 WASP-deficient monocytes were skewed toward an inflammatory phenotype with poor regulatory cytokine expression, driving colitis in a mouse model,46 and also, they were shown to have overactive inflammasomes and defective autophagy.47 A recent report has suggested that WASP-deficient platelets have an intrinsic capacity to drive normal B cells to produce autoantibodies owing to upregulation of activation markers and direct activation of B cells via CD40L-CD40.48
In contrast to correction of eczema and infections related to lack of T-cell function, correction of autoimmunity after HCT is less reliable, and patients remain at risk of recurrent or de novo autoimmunity after HCT.38,49 Two studies reported that patients with autoimmunity after HCT were more likely to have mixed chimerism.38,49 The complex cellular mechanisms of autoimmunity in WAS make it challenging to determine which lineage or lineages (Tregs, B cells, Bregs, myeloid cells, and platelets) are primarily responsible for autoimmunity post-HCT. The strong selective advantage of WASP-expressing T cells and Tregs over WASP-deficient cells and the presence of autoimmunity in mouse models deficient only in B cells argue against a failure of Treg reconstitution as a major driver of autoimmunity post-HCT. However, because Treg reconstitution from the thymus is expected to take 6 months or more, autoimmunity that occurs before substantial numbers of thymically derived T cells have been generated could potentially be attributed in part to failure of donor-derived Treg reconstitution. The intrinsic role of residual or persistently generated WASP-deficient B cells, myeloid cells, or platelets or a failure to reconstitute Breg in post-HCT autoimmunity in patients has yet to be fully explored. In the GT setting, rituximab was included in the conditioning regimen in the trial conducted in Milan in an attempt to stave off post-GT autoimmunity, and in the 9 patients treated, little to no autoimmunity has been seen.50 In other trials where ritixumab was not routinely given, autoimmunity post-GT has been seen (A. Thrasher, C. Booth, and S.-Y. Pai, Great Ormond Street Hospital, oral communication, 5 August 2019), and the cellular mechanism is being actively investigated.
Outcomes of HCT and GT for CGD
CGD is caused by mutations in genes encoding components of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex (Table 1), which is required for generation of bactericidal reactive oxygen species. In addition to bacterial and fungal infection, CGD patients suffer from granulomatous infiltration and hyperinflammation manifested as inflammatory bowel disease, lung disease, and obstruction of gastrointestinal or genitourinary tract, often requiring treatment with corticosteroids and other agents. The primary goal of HCT in CGD is thus to achieve high-level myeloid (neutrophil and monocyte) donor chimerism.
In contrast to historical experiences, recent outcomes of HCT for CGD have improved, in particular as reported in a multi-institutional study of 56 patients with CGD undergoing matched sibling donor or well-matched unrelated donor HCT with a uniform reduced intensity conditioning regimen of moderate-intensity pharmacokinetically adjusted busulfan, fludarabine, and serotherapy with either rabbit ATG or alemtuzumab.51 Despite a study population with a high proportion of patients of older age or high-risk features, survival was excellent, and only 5% of patients had graft failure. Furthermore, 93% of surviving patients achieved myeloid donor chimerism of ≥90%.
Because the various genetic causes of CGD interfere with neutrophil function but not with neutrophil development, there is no selective advantage for oxidase-expressing neutrophils, and therefore, the need for sufficient myeloablation is paramount. In contrast to SCID and WAS, where selective advantage of donor-derived T cells results in high-level donor T-cell chimerism even in states of mixed HSC chimerism, in CGD patients, high-level neutrophil chimerism may be accompanied by mixed T-cell chimerism. The PIDTC has conducted a retrospective review of 145 patients with CGD undergoing HCT and found that T-cell chimerism (range, 62% to 100%) was lower than myeloid chimerism (median, 100%).52
The minimum level of stable myeloid chimerism needed post-HCT to protect against CGD-related infections and/or hyperinflammation remains controversial. In X-linked CGD caused by mutations in the CYBB gene, female carriers exhibit a wide range of X-inactivation percentages in neutrophils. Patients with severely skewed lyonization favoring the mutant CYBB allele may suffer from all of the same complications as CGD patients. A study of 162 X-linked CGD carrier females suggested that neutrophil oxidase activity of >20% may be sufficient to protect against infections but not against immune dysregulation.53 Ascertaining the minimum level of normal myeloid cells needed for disease amelioration is critical for the design and interpretation of GT trials for CGD. GT trials for X-linked CGD using unmodified gammaretroviruses were plagued not only with insertional oncogenesis but also, transience of gene-marked neutrophils and loss of transgene function owing to gene silencing.54,55 A multi-institutional trial of a lentiviral vector expressing the CYBB gene under the control of a novel myeloid-specific chimeric promoter56 has shown preliminary evidence of efficacy despite partial reconstitution of oxidase activity, but it requires longer follow-up of clinical manifestations and durability of gene marking.57
Case evolution and conclusions
The boy with X-linked SCID would be offered allogeneic HCT with any of the alternative donor types available or autologous GT. Using a busulfan-based conditioning regimen (administered no earlier than 8 weeks of age) would be the best strategy to ensure full immune reconstitution with a single procedure.
The case illustrates the benefit of ascertainment of the genetic cause of PID, opening up the possibility for tailored therapy, including autologous GT. Head to head comparisons of HCT and GT are impractical in these rare patients; we must rely instead on detailed collection of immune and clinical outcome data, preferably in a uniform fashion across trials. In addition to the diseases discussed here, a large category of primary immune regulatory disorders is increasingly treated with HCT. Studies of genotype-specific outcomes of these disorders will be critical to determine which patients should proceed to cellular therapy and optimize their outcome.
Correspondence
Sung-Yun Pai, Boston Children's Hospital/Dana-Farber Cancer Institute, 300 Longwood Ave, Karp Family Research Laboratory, Room 08214, Boston, MA 02115; e-mail: sung-yun.pai@childrens.harvard.edu.
