
Abstract
The management of chronic lymphocytic leukemia (CLL) has undergone dramatic changes over the previous 2 decades with the introduction of multiple new therapies and new combinations. Management of the newly diagnosed asymptomatic patient has not significantly changed outside of the development of a number of prognostic factors and the CLL International Prognostic Index, which is helpful in discussions regarding prognosis and likelihood of requiring treatment. When therapy is required, initial treatment of most patients now includes either the Bruton tyrosine kinase inhibitor ibrutinib or the B-cell lymphoma 2 inhibitor venetoclax in combination with obinutuzumab. Current frontline trials are focused on the optimal sequencing or combination of targeted therapies. In this review, we will discuss the management of previously untreated CLL with an emphasis on the clinical trials that have formed the standard of care, as well as those newer studies that are likely to form the next generation of therapy.
Learning Objectives
Discuss the management of newly diagnosed CLL
Discuss the clinical trials that formed the basis of current frontline treatment in CLL
Clinical case
Your patient is a 68-year-old man with newly diagnosed chronic lymphocytic leukemia (CLL). He was diagnosed by his primary care physician, who had drawn blood at a routine visit and found that the patient had a lymphocytosis with an absolute lymphocyte count of 21 000. Peripheral blood flow cytometry was diagnostic for CLL with immunophenotype CD5+/CD19+/CD20+(dim)/CD23+/CD79b−. He is referred to you for management. On history and physical examination, you determine that he has no symptoms attributable to his disease and no palpable lymph nodes or splenomegaly. Hemoglobin and platelet count are normal. Because the patient does not meet criteria for the initiation of therapy, further prognostic testing is not mandated, and in many cases is not preferred because certain studies would need to be repeated at the time of therapy initiation. However, the patient is interested in better understanding the genetic risk of his disease and overall prognosis, so you collect blood for immunoglobulin heavy chain (IGHV) mutational status, β2 microglobulin (B2M), fluorescence in situ hybridization (FISH) testing, and TP53 mutation. These return showing unmutated IGHV, normal B2M, del(11q22.3), and no evidence of TP53 mutation. Using the CLL International Prognostic Index (IPI), you determine that he has intermediate-risk disease, with a 5-year survival probability of 79.4%.
He asks you when he is going to require therapy, and whether there is a role for early treatment…
Treatment-naive CLL: lessons from phase 2 and phase 3 clinical trials
The management of CLL has evolved dramatically over the past 2 decades, due to both advancement of therapeutics as well as increased understanding of the disease biology, allowing for development of relevant prognostic markers. In this article, I will discuss the initial management of CLL, with a focus on the definitive clinical trials that have defined the current standards of care.
CLL is an extremely heterogenous disease for which some patients will need therapy very close to the time of their initial diagnosis and will receive multiple therapies during the course of their life; others may never require treatment. In CLL, a number of prognostic factors have been identified that can be extremely helpful when counseling patients. The earliest prognostic factors, the Rai1 and Binet2 staging systems, remain in widespread use. Although the survival estimates with these systems are no longer accurate given the advances in therapy, it still very likely remains true that those patients with higher stage at diagnosis have shorter survival than those at earlier stages. In addition to clinical stage, cytogenetic and molecular markers can add greatly to prognostication. FISH came into widespread use almost 2 decades ago. As sole abnormalities, the presence of del(13q14.3) imparts favorable prognosis, trisomy 12 confers intermediate risk, and del(11q22.3) and del(17p13.1) are associated with poor prognosis.3 When multiple abnormalities exist, the impact on prognosis is determined by the following hierarchy: del(17p13.1) > del(11q22.3) > trisomy12 > del(13q14.3). del(17p13.1) is associated with loss of the TP53 tumor suppressor on 1 chromosome. In ∼80% of cases, this coexists with a mutation in the alternate allele of TP53, although mutations (often with a dominant-negative phenotype) can occur in isolation.4-6 TP53 disruptions through loss or mutations are associated with similarly poor outcomes. Stimulated karyotype can also add to prognosis in some cases, with a complex karyotype (≥3 abnormalities) associated with a more aggressive disease course.7,8 Cytogenetics and mutations can change over time, most commonly following therapy, with acquisition of new abnormalities and an increasing karyotypic complexity, a process known as clonal evolution.9-11 Another strong predictor of disease course is the degree of somatic hypermutation of the variable region of the IGHV. Somatic hypermutation (>2% difference from germline) may indicate a post–germinal center derived progenitor cell and is associated with a much more indolent course than unmutated CLL. Patients with mutated CLL are less likely to require therapy, respond better to chemotherapy, and have prolonged survival compared with those with unmutated IGHV.12,13 Unlike karyotype, IGHV mutational status does not change over time.
The CLL IPI was developed to integrate the major prognostic factors in CLL into an index that would be helpful in clinical practice. It takes into account 5 independent prognostic factors: age, clinical stage, IGHV mutational status, TP53 abnormalities, and B2M, and separates patients into 4 distinct risk groups. These risk groups have significantly different survival at 5 years, from 93.2% 5-year survival in the low-risk group to 23.3% in the very high-risk group. It is important to take into account, however, when using any of the established systems to estimate survival, that none of these were developed in the era of kinase inhibitor treatment of CLL, and very likely all underestimate the actual survival in this disease. Although initially designed to predict survival, the CLL IPI can also be used to calculate time to first treatment, as can the MD Anderson Cancer Center (MDACC) score14 and the O-CLL1 score.15 External validation has shown similar accuracy among these calculators,16 although none can perfectly assess risk for an individual patient. The CLL IPI can easily be calculated in clinical practice, and multiple online calculators exist.
Your patient’s prognostic studies indicate he has intermediate-risk disease. The CLL IPI would estimate a 79.4% chance of 5-year survival and a time to first treatment of about 4 years from diagnosis. CLL is currently treated at the onset of marrow failure or symptoms related to disease, as established by the International Working Group on CLL (iwCLL), so this patient would not currently meet criteria to initiate therapy.17 Symptoms/signs that merit consideration of therapy can be found in Table 1, and briefly include symptomatic lymphadenopathy or splenomegaly, cytopenias related to bone marrow infiltration, autoimmune complications not responsive to standard treatment, and constitutional symptoms due to disease, most commonly fatigue. This strategy was established based upon a number of randomized phase 3 trials that have not demonstrated a survival benefit for early treatment. The first of these studies randomized patients who were newly diagnosed and asymptomatic to observation vs single-agent chlorambucil, and found no difference in overall survival (OS) between the 2 groups.18 Limitations of this study include relatively ineffective therapy in chlorambucil, and no knowledge at that time of prognostic factors beyond clinical stage. With increasing understanding of molecular prognostic markers as well as more effective treatment, a phase 3 study was undertaken that first stratified asymptomatic untreated patients to low- vs high-risk disease, and then randomized those with high-risk disease to fludarabine/cyclophosphamide/rituximab (FCR) vs observation.19 Although there was an improvement in event-free survival for patients receiving FCR, there was no survival advantage to early treatment vs observation. Notably, there was a significant difference in both event-free survival and OS for low-risk patients compared with high-risk patients regardless of therapy, emphasizing the importance of these prognostic factors in the management of CLL.
Criteria for the initiation of therapy in CLL
| Criteria for the initiation of therapy in CLL (adapted from iwCLL 2018 guidelines) |
|---|
| Marrow failure: hemoglobin <10 g/dL or platelet count <100 × 109/L |
| Massive, progressive, or symptomatic splenomegaly |
| Massive, progressive, or symptomatic lymphadenopathy |
| Progressive lymphocytosis with ≥50% increase over 2-mo period, or lymphocyte doubling time <6 mo |
| Autoimmune complications poorly responsive to corticosteroids |
| Symptomatic extranodal involvement |
| Disease-related symptoms: weight loss ≥10% over 6 mo, significant fatigue, fevers for 2 or more weeks without infection, night sweats ≥1 mo without infection |
| Criteria for the initiation of therapy in CLL (adapted from iwCLL 2018 guidelines) |
|---|
| Marrow failure: hemoglobin <10 g/dL or platelet count <100 × 109/L |
| Massive, progressive, or symptomatic splenomegaly |
| Massive, progressive, or symptomatic lymphadenopathy |
| Progressive lymphocytosis with ≥50% increase over 2-mo period, or lymphocyte doubling time <6 mo |
| Autoimmune complications poorly responsive to corticosteroids |
| Symptomatic extranodal involvement |
| Disease-related symptoms: weight loss ≥10% over 6 mo, significant fatigue, fevers for 2 or more weeks without infection, night sweats ≥1 mo without infection |
Clinical case revisited
Five years later, after 2 years of slow disease progression, your patient presents with a lymphocyte count of 126, a hemoglobin count of 10.6 g/dL, and a platelet count of 89 × 109. His performance status remains excellent, and he spends most days working in his yard or playing golf. You perform a bone marrow biopsy, which shows >90% CLL in the marrow, with decreased normal hematopoiesis. There is no evidence of myelodysplasia in the marrow. There are also no signs of hemolysis, consisting of a negative direct Coombs test, normal lactate dehydrogenase and bilirubin, and decreased reticulocyte count. Kidney and liver function is normal, with a glomerular filtration rate of 80 mL/min and liver function tests within normal limits. Based on iwCLL guidelines, you decide to initiate therapy. Prior to starting treatment, you repeat FISH and TP53 mutation testing, which redemonstrates del(11q22.3) and is again negative for TP53 mutations. Your practice does not conduct clinical trials, and your patient expresses interest in being treated locally with the most effective standard-of-care therapy. You think about the best option for treatment…
Continued lessons from phase 2 and phase 3 clinical trials
At 73 years of age with a good performance status and minimal comorbidities, your patient is the definition of an older, fit CLL patient. His therapy should therefore optimally extend his life while preserving his fitness and quality of life. Randomized phase 3 studies that have formed standard of care for symptomatic, previously untreated CLL are found in Table 2. In the past, chemoimmunotherapy (CIT) was the best option for patients with CLL. Although fludarabine-based CIT was standard initial therapy for patients younger than 65 to 70 years of age, optimal initial therapy for older adults with CLL was not as well established. The CLL11 trial established chlorambucil plus obinutuzumab as the most appropriate standard of care for unfit CLL patients, however, with a median progression-free survival (PFS) of only 26.7 months,20 it does not appear to be as effective as the more aggressive CIT regimens for fit patients. A phase 3 study of FCR vs bendamustine/rituximab (BR) showed that BR was inferior to FCR, except in the subset of patients older than 65 years, where the 2 treatments appeared similarly effective.21 These data, as well as phase 2 data22 and the phase 3 MABLE study (which showed that BR was superior to chlorambucil/rituximab with an overall response rate [ORR] of 91% and median PFS of 40 months23 ), established BR as a standard of care, which was widely adopted. Recently though, the movement of the Bruton tyrosine kinase (BTK) inhibitor ibrutinib into the frontline setting has begun to change the paradigm of treatment of CLL. The most mature data in the frontline setting with ibrutinib comes from PCYC 1102, a phase 1b/2 study of single -agent ibrutinib administered indefinitely,24 in which 31 patients were treated in the upfront setting. ORR, which deepened over time, was 89%, with 29% of patients achieving a complete response (CR). At 5 years, 92% of patients were alive and free of progression.25 Ibrutinib received US Food and Drug Administration (FDA) approval in the frontline setting based on the RESONATE 2 trial, which randomized previously untreated patients age 65 years and older to ibrutinib or chlorambucil. At 18 months, ibrutinib decreased the risk of progression or death by 84% compared with chlorambucil.26 At more recent follow-up of 29 months, ORR is 92% and 2-year PFS is 88%.27
Relevant phase 3 clinical trials in CLL
| Trial | Regimens compared | ORR, % | PFS | OS | Results |
|---|---|---|---|---|---|
| CLL831 | FC | 80 | Median, 32.9 mo | Median, 86 mo | FCR is superior to FC in terms of PFS |
| FCR | 90 | Median, 56.8 mo | Median, not reached | FCR is superior to FC in terms of PFS | |
| CLL1021 | FCR | 95 | Median, 55.2 mo | 3-y, 91% | BR is inferior to FCR in terms of PFS |
| BR | 96 | Median, 41.7 mo | 3-y, 92% | ||
| CLL1120 | Ch | 65.7 | Median, 11.1 mo | NR | ChO is superior to Ch and ChR in terms of PFS and OS |
| ChR | 31.4 | Median, 15.4 mo | NR | ||
| ChO | 77.3 | Median, 29.2 mo | NR | ||
| RESONATE 226 | Ch | 35 | 24-mo, 34% | 24-mo, 84% | Ibrutinib is superior to Ch in terms of PFS and OS |
| Ibrutinib | 92 | 24-mo, 89% | 24-mo, 95% | ||
| E191234 | IR | 96 | 3-y, 89% | 3-y, 99% | IR is superior to FCR in terms of PFS and OS |
| FCR | 81 | 3-y, 73% | 3-y, 92% | ||
| A04120228 | BR | 81 | 2-y, 74% | 2-y, 95% | Ibrutinib and IR are superior to BR in terms of PFS, IR is no more effective than ibrutinib in terms of PFS |
| Ibrutinib | 93 | 2-y, 87% | 2-y, 90% | ||
| IR | 94 | 2-y, 88% | 2-y, 94% | ||
| iLLUMINATE30 | ChO | 73 | 30-mo, 31% | 30-mo, 85% | IO is superior to ChO in terms of PFS |
| IO | 88 | 30-mo, 79% | 30-mo, 86% | ||
| CLL1435 | ChO | 71.3% | 2-y, 64.1% | 2-y, 93.3% | VO is superior to ChO in terms of PFS |
| VO | 84.7% | 2-y, 88.2% | 2-y, 91.8% |
| Trial | Regimens compared | ORR, % | PFS | OS | Results |
|---|---|---|---|---|---|
| CLL831 | FC | 80 | Median, 32.9 mo | Median, 86 mo | FCR is superior to FC in terms of PFS |
| FCR | 90 | Median, 56.8 mo | Median, not reached | FCR is superior to FC in terms of PFS | |
| CLL1021 | FCR | 95 | Median, 55.2 mo | 3-y, 91% | BR is inferior to FCR in terms of PFS |
| BR | 96 | Median, 41.7 mo | 3-y, 92% | ||
| CLL1120 | Ch | 65.7 | Median, 11.1 mo | NR | ChO is superior to Ch and ChR in terms of PFS and OS |
| ChR | 31.4 | Median, 15.4 mo | NR | ||
| ChO | 77.3 | Median, 29.2 mo | NR | ||
| RESONATE 226 | Ch | 35 | 24-mo, 34% | 24-mo, 84% | Ibrutinib is superior to Ch in terms of PFS and OS |
| Ibrutinib | 92 | 24-mo, 89% | 24-mo, 95% | ||
| E191234 | IR | 96 | 3-y, 89% | 3-y, 99% | IR is superior to FCR in terms of PFS and OS |
| FCR | 81 | 3-y, 73% | 3-y, 92% | ||
| A04120228 | BR | 81 | 2-y, 74% | 2-y, 95% | Ibrutinib and IR are superior to BR in terms of PFS, IR is no more effective than ibrutinib in terms of PFS |
| Ibrutinib | 93 | 2-y, 87% | 2-y, 90% | ||
| IR | 94 | 2-y, 88% | 2-y, 94% | ||
| iLLUMINATE30 | ChO | 73 | 30-mo, 31% | 30-mo, 85% | IO is superior to ChO in terms of PFS |
| IO | 88 | 30-mo, 79% | 30-mo, 86% | ||
| CLL1435 | ChO | 71.3% | 2-y, 64.1% | 2-y, 93.3% | VO is superior to ChO in terms of PFS |
| VO | 84.7% | 2-y, 88.2% | 2-y, 91.8% |
BR, bendamustine/rituximab; Ch, chlorambucil; ChO, chlorambucil/obinutuzumab; ChR, chlorambucil/rituximab; FC, fludarabine/cyclophosphamide; FCR, fludarabine/cyclophosphamide/rituximab; IO, ibrutinib/obinutuzumab; IR, ibrutinib/rituximab; iwCLL, International Working Group on CLL; NR, not reported; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
The RESONATE 2 study has been criticized for the lack of a standard-of-care control arm, although it can certainly be argued that there was no established gold standard for this age group. More recently, A041702, an Alliance-led National Clinical Trials Network (NCTN) study, compared ibrutinib to ibrutinib/rituximab (IR) and BR. With median follow-up of 38 months, both ibrutinib and IR showed superior PFS to BR, with a hazard ratio of 0.38 for ibrutinib and 0.39 for IR.28 There was no significant difference between ibrutinib and IR, with a hazard ratio of 1.00. At 2 years, PFS was 74% (95% confidence interval [CI], 66% to 80%) with BR, 87% (95% CI, 81% to 92%) with ibrutinib, and 88% (95% CI, 81% to 92%) with IR. OS was no different between the arms. Hematologic toxicities were more common with BR, whereas nonhematologic toxicities in general were slightly more common with ibrutinib, primarily driven by toxicities known to be associated with the drug, including atrial fibrillation and hypertension. These toxicities, especially atrial fibrillation (seen in ∼15% of patients) and ventricular arrhythmias (seen much more rarely),29 are a focus of ongoing research and do need to be considered when choosing therapy, especially in patients with cardiac comorbidities. However, these data demonstrate that for older treatment-naive patients, ibrutinib should be considered a, if not the, standard of care. Another recent study comparing ibrutinib-based regimens to CIT in the frontline setting primarily for older patients is the iLLUMINATE study, which randomized previously untreated patients who were 65 years or older, or younger than 65 years but with comorbidities, to ibrutinib/obinutuzumab (IO) vs chlorambucil/obinutuzumab. At a median follow-up of 31.3 months, median PFS was 19.0 months in the chlorambucil/obinutuzumab arm and not reached for IO.30 Thirty-month PFS was 79% (95% CI, 70% to 85%) for IO and 31% (95% CI, 23% to 40%) for chlorambucil/obinutuzumab. Although more patients were in CR in this study compared with A041202 (19% vs 7%), it is difficult to know whether obinutuzumab offers an advantage over ibrutinib alone because rituximab does not.
For younger patients as well, the paradigm for treatment-naive disease has recently switched from CIT to targeted therapy based upon the recently published E1912 study. Prior to this study, FCR was the clear standard of care for young, fit patients based upon the CLL8 and CLL10 studies. The CLL8 study randomized patients to FCR vs fludarabine/cyclophosphamide (FC), and showed both PFS and OS advantages with FCR,31,32 with a median PFS of 56.8 months for FCR vs 32.9 months for FC. Additionally, the CLL10 study showed that BR was inferior to FCR in younger fit patients.21 Most compelling with FCR is the accumulating evidence that a significant subset of patients with good-risk disease (mutated IGHV, no del17p) will have prolonged PFS and potentially a cure with this regimen.32,33 E1912 randomized patients younger than 70 years of age to FCR vs IR. At a median follow-up of 33.6 months, IR has significantly longer PFS than FCR, with a hazard ratio of 0.35 for the intention-to-treat patient population.34 Unexpectedly, given the short follow-up, OS also favored IR, with a hazard ratio of 0.17. The results are primarily driven by the IGHV -unmutated subset of patients, whereas in the mutated subset, very few patients have relapsed. Therefore, long-term follow-up of the study will be critically important to determine whether, in these very good-risk patients with potential for cure, IR remains superior to FCR. Notably, in this study, atrial fibrillation was seen less commonly than in the Alliance study (7.4%); however, because the drug will be given indefinitely, it seems likely that this number will increase as the study population ages, again highlighting the importance of considering potential cardiac complications of ibrutinib when choosing therapy.
Most recently, the CLL14 trial randomized patients with previously untreated CLL and comorbid conditions to treatment with venetoclax/obinutuzumab vs chlorambucil/obinutuzumab. Median age on the study was 72 years. In contrast to BTK inhibitors, for which therapy is indefinite, both treatment regimens were given for a duration of 1 year. At a median follow-up of 28.1 months, 24-month PFS was 88.2% in the venetoclax/obinutuzumab arm compared with 64.1% in the chlorambucil/obinutuzumab arm.35 OS was not different between the arms. Long-term follow-up will be very helpful to determining the length of remissions after treatment as well as the efficacy of retreatment, but this is an ideal approach for a patient who does not want to commit to long-term treatment with a BTK inhibitor. For select patients with low-risk disease, CIT can also be considered in the context of patients wishing to delay initiation of indefinite therapy.
These data from randomized phase 3 trials have significantly impacted therapeutic decision making in frontline CLL. Additionally, although patients with del(17p13.1) deletion or TP53 mutation continue to have outcomes that are inferior to those with intact TP53, the success of BTK inhibitors and venetoclax in this context has resulted in the same initial treatment regimens preferred for patients with or without TP53 abnormalities. Figure 1 demonstrates a reasonable algorithm for the initial therapy of CLL.
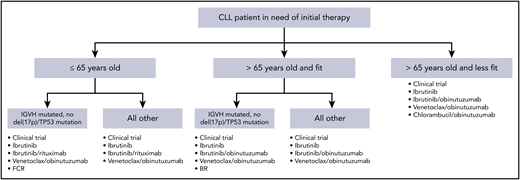
Algorithm for initial therapy of CLL. This is a suggested algorithm to approach CLL patients in need of therapy adapted from the NCCN (www.nccn.org) and Onkopedia (www.onkopedia.com) guidelines. Options are not listed in order of preference. Clinical trials should be considered for patients for whom this option is available.
Algorithm for initial therapy of CLL. This is a suggested algorithm to approach CLL patients in need of therapy adapted from the NCCN (www.nccn.org) and Onkopedia (www.onkopedia.com) guidelines. Options are not listed in order of preference. Clinical trials should be considered for patients for whom this option is available.
Clinical case posttreatment
After a discussion of the options for frontline therapy outside of clinical trials, your patient is started on ibrutinib and has an excellent response. He has an initial lymphocytosis, which is expected, but over the course of a year of treatment his white blood cell count slowly decreases. He has joint aches, which are able to be controlled with acetaminophen and occasional use of ibuprofen. One initial flare of these joint aches was treated with low-dose prednisone for 2 weeks, which greatly improved his symptoms. At 1 year of treatment, he has no palpable lymph nodes, and peripheral blood counts have normalized. Flow cytometry continues to reveal >50% CLL cells in the peripheral blood, which is expected. During 1 of his clinic visits, he asks you whether it is necessary that he continue on ibrutinib indefinitely, and wonders if there would be other options…
More lessons from phase 2 and phase 3 clinical trials
Since the first clinical trials of ibrutinib, it has been assumed that this agent needs to be continued indefinitely for efficacy. Although responses rates with ibrutinib are extremely high and remissions durable, CRs are uncommon, even in the frontline setting. In A041202, the CR rate after a median duration of almost 3 years is only 7%, and minimal residual disease (MRD) negativity in the bone marrow was only seen in 1% of patients after 9 cycles of treatment. This would suggest that therapy would need to be continued without interruption to prevent disease progression. Discontinuation of therapy, if effective, would have a number of advantages. First, the cost of the drug is extremely high, and treatment-free intervals would relieve some financial burden on patients as well as the health care system. Second, discontinuation would prevent treatment-related toxicities. Although many patients tolerate ibrutinib well, and there are patients from the original clinical trials with this agent who have been receiving continuous therapy for >8 years, there are significant complications that can occur with this agent that make intermittent therapy more appealing if feasible. These include uncommon but serious events such as atrial fibrillation and bleeding, as well as more common lower grade toxicities that are very bothersome when persistent, such as joint or muscle pain. Additionally, hypertension is increasingly recognized as a complication of ibrutinib, and the impact of this on long-term cardiovascular health in patients on ibrutinib is unknown.
A “real-world” analysis of ibrutinib from multiple institutions showed that ibrutinib is not as well tolerated outside of clinical trials, with a significant proportion of patients discontinuing therapy due to toxicity.36 At a median follow-up of 17 months, 41% of patients discontinued therapy. Of those patients in the frontline setting who discontinued, 63% did so because of toxicity, most commonly arthralgia, atrial fibrillation, and rash. These data further emphasize that identifying strategies for patients to successfully discontinue ibrutinib is of high interest.
The most promising strategy at this time for ibrutinib discontinuation involves combinations with the oral B-cell lymphoma 2 (BCL2) inhibitor venetoclax, with or without obinutuzumab (ibrutinib/venetoclax [IV] and ibrutinib/venetoclax/obinutuzumab [IVO], respectively). Two large phase 2 studies of the doublet have been conducted: the Bloodwise TAP CLARITY trial in relapsed CLL and a phase 2 study from MD Anderson in the frontline setting. In the CLARITY study, which included 50 patients, the response rate was 100% after 12 months of therapy. Fifty-eight percent of patients achieved a CR, and 41% had undetectable MRD in the bone marrow.37 The MD Anderson trial included 80 patients, 92% of whom had high-risk disease, defined as IGVH unmutated disease, TP53 abnormality, or del(11q22.3).38 Patients received 24 cycles of IV; at 12 months, 92% of patients were in CR or CR with incomplete marrow recovery, and 68% of patients had undetectable MRD in the bone marrow. The triplet of IVO has been studied in a phase 1b/2 trial at The Ohio State University. Twelve patients were enrolled in the phase 1b setting, and then 50 in the phase 2 (25 relapsed, 25 treatment-naive), with patients receiving a total of 12 months of treatment. Among the patients with treatment-naive disease, 92% achieved a response, and 28% achieved an MRD− CR. Undetectable MRD was observed in the bone marrow of 67% of patients.39 Although the responses for IV and IVO are exceptionally high, none of these trials has the follow-up needed to determine whether discontinuation of therapy in this case will be successful. Therefore, neither of these regimens should be used outside of the context of a clinical trial. Two NCTN phase 3 clinical trials are ongoing that will determine whether combination therapy with IVO is superior to IO in frontline CLL (A041702 for older patients and EA9161 for younger patients).
Conclusions
The therapy of CLL has undergone significant changes over the previous 2 decades, and is poised to continue to evolve. The recent introduction of targeted therapies has changed the prognosis for patients, especially those with high genomic-risk disease, and newer combination studies hold hope for continued progress. The improvements in therapy for patients with CLL has been phenomenal and provide hope for both continued breakthroughs in this disease as well as other malignancies. In addition to the development of novel agents, this success has been achieved by the dedication of physicians and patients who have participated in the clinical trials that have introduced these new treatments. A cure for all patients has yet to be achieved, though, emphasizing the importance of continued participation in these large randomized clinical trials that ultimately determine optimal standard of care. This is an exciting time in CLL, with all evidence pointing toward continued improvements in the care of our patients with this disease.
Correspondence
Jennifer A. Woyach, The Ohio State University, 455D Wiseman Hall CCC, Columbus, OH 43221; e-mail: jennifer.woyach@osumc.edu.
