
Abstract
The landscape of acute lymphoblastic leukemia (ALL) has evolved significantly over the last few years. Identification of specific recurrent genetic alterations and of minimal residual disease (MRD) guides prognostic classification and management. Novel agents (eg, blinatumomab) have demonstrated encouraging results in relapsed/refractory (R/R) and MRD+ patients and are currently incorporated into upfront treatment in specific settings. Other new strategies include the incorporation of tyrosine kinase inhibitor-based therapy for patients with Philadelphia chromosome–like ALL and the use of DOT inhibitors and bcl-2/bcl-xl inhibitors in R/R disease. These innovations promise to improve management and outcome in this disease.
Learning Objectives
Describe the role of genetic alterations, including those associated with Philadelphia chromosome–like acute lymphoblastic leukemia (ALL) in determining treatment strategies
Discuss the prognostic impact of minimal residual disease
Review outcomes following blinatumomab and inotuzumab in the treatment of ALL
Explain the importance of the bcl-2/bcl-xl pathway in ALL and inhibitors of that pathway in ongoing clinical trials
Clinical case
Patient B is a 56-year-old man with newly diagnosed B-cell acute lymphoblastic leukemia (B-ALL). His past medical history is notable for hypertension and hyperlipidemia. Laboratory results on admission include white blood cells (WBCs), 0.8 K/μL; hemoglobin (Hgb), 7 g/dL; and platelets (Plts), 20 K/μL. Bone marrow aspirate/biopsy demonstrates 90% blasts and flow cytometry is CD19+, CD20−, CD22+, CD34+, CD38+, CD45+, HLA-DR+, terminal deoxynucleotidyl transferase (TdT+), diagnostic for B-ALL. Cytogenetics are 46 XY; t(1;4) (q32; q35); +11, −20. Reverse-transcriptase polymerase chain reaction (RT-PCR) for Bcr-abl is negative. Testing for the Philadelphia (Ph)-like signature is negative for any ABL class fusions or JAK pathway alterations. The patient consents to and is enrolled on E1910, a randomized phase 3 clinical trial of chemotherapy with or without blinatumomab.
Introduction
The evaluation and management of this patient incorporates many of the advances in our approach to acute lymphoblastic leukemia (ALL) made over the last decade. We will use this case to discuss: (1) the prognostic and therapeutic impact of recurrent genetic abnormalities in ALL, (2) the prognostic impact of minimal residual disease (MRD), and (3) the incorporation of novel therapies in the treatment of ALL (Figure 1). We will focus primarily on selected influential advances in B-ALL, but will not discuss chimeric antigen receptor (CAR) T-cell therapy because this is a separate chapter on its own.
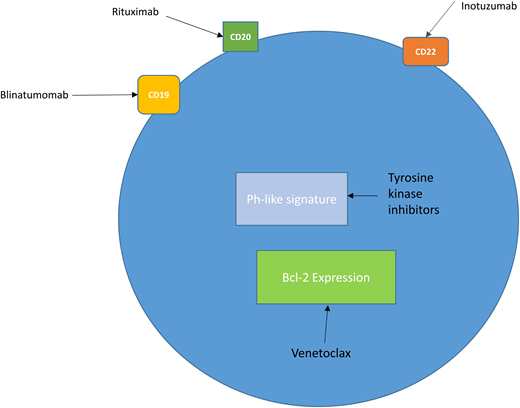
New treatment strategies in ALL. B lymphoblasts express the cell surface antigens CD19, CD20, and CD22, which are targets for the antibody therapies blinatumomab, rituximab, and inotuzumab. A subset of patients with B-ALL (20%-25%) express the Ph-like signature. These patients may be candidates for clinical trials with TKI–based therapy. Bcl-2 is overexpressed in both B- and T-ALL, and current trials are evaluating the bcl-2 inhibitor venetoclax in R/R disease.
New treatment strategies in ALL. B lymphoblasts express the cell surface antigens CD19, CD20, and CD22, which are targets for the antibody therapies blinatumomab, rituximab, and inotuzumab. A subset of patients with B-ALL (20%-25%) express the Ph-like signature. These patients may be candidates for clinical trials with TKI–based therapy. Bcl-2 is overexpressed in both B- and T-ALL, and current trials are evaluating the bcl-2 inhibitor venetoclax in R/R disease.
Recurring genetic alterations
Although early classifications of ALL were based on morphology, and risk stratification on age and WBC count, recurrent genetic abnormalities have now become critical determinants of prognosis and management. Risk stratification identifies patients at high risk of relapse with standard therapeutic approaches who may benefit from more intensive treatment including hematopoietic cell transplantation (HCT). Adverse genetic abnormalities include the Ph chromosome, t(4;11)/KMT2A-AFF1 rearrangement, low hypodiploidy, mutated TP53 and Ph-like ALL.1 The frequency, and historically poor survival, of Ph+ disease is responsible for its prominent impact on therapeutic approach and outcome. Incorporation of tyrosine kinase inhibitors (TKIs) has improved outcomes of Ph+ ALL immensely and serves as a model for targeted approaches as a part of individualized treatment strategies in this heterogeneous disease.
MLL rearrangements
Mixed lineage leukemia (MLL; KMT2A) rearrangements occur in ∼5% of patients and confer a poor prognosis. They are associated with increased H3K79 methylation catalyzed by DOTL1. DOT inhibitors demonstrate antiproliferative activity in MLL-rearranged leukemia cells and are being studied in clinical trials in relapsed/refractory (R/R) cases.2 MLL rearrangement also results in high expression of bcl-2, BAX, and BIM, leading to susceptibility of ALL to the bcl-2 inhibitor venetoclax.3,4
Ph-like signature
The Ph-like signature was first identified in children with poor outcomes in whom gene-expression profiling identified a “kinase”-like signature reminiscent of Ph+ ALL.5 The incidence of Ph-like ALL is ∼10% to 15% in children, ∼25% in adolescents and young adults (AYAs), and ∼20% to 30% in adults.6-9 Kinase-activating alterations have been identified in 91% of these patients and include rearrangements involving ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK2, NTRK3, PDGFRB, PTK2B, TSLP, or TYK2 and mutations involving FLT3, IL7R, or SH2B3.10 The major kinase alterations observed in adults are: 51% CRLF2 rearrangements, 12% JAK2 or EPOR rearrangements, 10% ABL class fusions, 7% JAK-STAT sequence mutations, 4% other kinase alterations, and 4% Ras pathway mutations.7 Initial trials demonstrated 5-year event-free survival (EFS) for young adults, adolescents, and high-risk children with Ph-like ALL of 24.1% ± 10.5%, 41.0% ± 7.4%, and 58.2% ± 5.3%, respectively.10 Adult ALL patients (ages 21-86 years) demonstrated inferior 5-year EFS compared with non–Ph-like ALL (22.5% vs 49.3%; P < .001).7 Many Ph-like patients are refractory to initial chemotherapy or have MRD detected postinduction,11 and poor outcomes regardless of MRD status have been reported.8 Thus, many experts recommend early HCT despite the absence of prospective evidence of its effectiveness. Many of the alterations may be sensitive to TKIs (Figure 2). Preclinical studies in vitro and in xenograft models demonstrate the efficacy of the ABL inhibitor dasatinib in ABL class fusions (ABL1, ABL2, CSF1R, PDGFRB) and of the JAK1/2 inhibitor ruxolitinib in JAK-STAT–activating alterations (JAK1, JAK2, JAK3, IL7R, IL2RB).12 Ongoing trials within the Children’s Oncology Group (COG) AALL1131 test all high risk patients for the Ph-like signature and then use combination therapy with dasatinib (for patients with ABL class fusions) or with ruxolitinib (AALL1521) for patients with JAK pathway alterations. Tasian reported constitutive and/or cytokine-inducible PI3K/AKT/mammalian target of rapamycin (mTOR) signaling in CRLF2-rearranged ALL.13 Preclinical studies demonstrate enhanced efficacy when combining the dual PI3K/ mTOR inhibitor gedatolisib with JAK or ABL inhibitors in murine xenograft models of Ph-like ALL.13
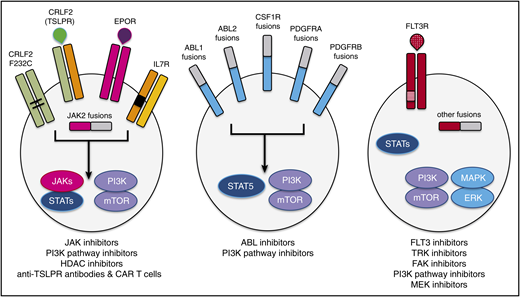
Schema of activated kinase signaling in Ph-like ALL. Kinase fusions and other alterations in Ph-like ALL activate oncogenic signal transduction and may be targetable by specific kinase inhibitors and other therapeutic agents. Reprinted from Tasian et al53 with permission.
Schema of activated kinase signaling in Ph-like ALL. Kinase fusions and other alterations in Ph-like ALL activate oncogenic signal transduction and may be targetable by specific kinase inhibitors and other therapeutic agents. Reprinted from Tasian et al53 with permission.
Assays for detection of Ph-like ALL vary and testing is not yet routine. More limited, targeted approaches are typically used in single-institution settings where flow cytometry is performed to evaluate overexpression of CRLF2 and fluorescence in situ hybridization (FISH) to detect rearrangement of CRLF2. For other known kinases (ABL1, ABL2, CSF1R, and JAK2), a break-apart kinase assay to assess for rearrangements can be performed.14 A comprehensive approach uses DNA and RNA sequencing, which detects all fusions and structural aberrations; however, this is expensive and labor intensive. A tiered approach used in current COG trials applies a quantitative RT-PCR–based low-density array platform to calculate a Ph-like coefficient of 0 to 1 based on the expression of 8 to 15 genes that were highly expressed in Ph-like ALL in a large validation cohort from AALL0232.14,15 Patients with a coefficient >0.5 are designated Ph-like ALL (Figure 3). Our current recommendation is to test patients upfront for the presence of the Ph-like signature (we send to Nationwide Children’s in Columbus, OH). If the patient has a Ph-like signature, they should be offered a clinical trial if one is available. This could include a trial with chemotherapy plus TKI (targeting the appropriate fusion) or a trial with chemotherapy plus one of the new antibodies (blinatumomab or inotuzumab). Given the absence of prospective data, the role of HCT is controversial, however, poor outcomes regardless of MRD from studies14 and in preliminary data from COG led many experts to recommend HCT in first complete remission (CR).
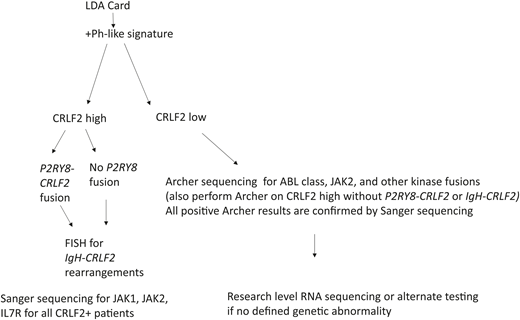
Algorithm for diagnosing the Ph-like signature being used in current COG trials. Information from Roberts.14
Algorithm for diagnosing the Ph-like signature being used in current COG trials. Information from Roberts.14
Back to our case…
Our patient is Ph− and Ph-like negative, however, he has 3 different cytogenetic alterations. Complex karyotype has been generally defined as ≥3 unrelated chromosomal abnormalities and is a well-established significant prognostic factor in AML. Its impact has been less well studied in ALL. Moorman et al demonstrated an adverse influence of 5 or more unrelated chromosomal abnormalities in adult ALL in the MRC UKALL XII/ECOG E2993 trial,16 and, although it has often been included as an adverse risk factor in adult ALL, its significance has not been validated in other large series, including 631 evaluable patients treated according to the risk-adapted protocols of the Programa Español de Tratamientos en Hematología (PETHEMA) group.17 Similarly, monosomal karyotype, defined as 2 or more autosomal monosomies or a single monosomy in the presence of structural abnormalities, is an unfavorable prognostic factor in AML but has been studied in only a limited fashion in ALL. The PETHEMA study detected no adverse impact of monosomal karyotype on overall survival (OS) or EFS.
The patient in our case thus did not demonstrate high-risk karyotype and was Ph-like negative. He receives standard induction on trial. Bone marrow postinduction is MRD− by multicolor flow cytometry performed in a centralized laboratory. Patient B completes intensification and is randomized to the blinatumomab arm. During his induction and postremission therapy course, he continues to receive prophylactic intrathecal chemotherapy. A decision is made not to proceed to transplant given his MRD− status and lack of high-risk karyotype.
Role of MRD
MRD is a critical prognostic factor in ALL and helps guide therapeutic decisions including decisions regarding transplantation. Multiple studies have confirmed the prognostic significance of MRD in children with ALL, and this has become an integral part of risk stratification and therapeutic decisions in pediatric trials.18-22 Berry et al22 examined MRD data from 13 637 pediatric and adult patients from 39 studies. Ten-year EFS for MRD− vs MRD+ patients was 77% vs 32% for pediatric patients and 64% vs 21% for adults. Bruggemann demonstrated the prognostic impact of MRD in 105 adult ALL patients treated uniformly with chemotherapy.18 MRD was measured by quantitative polymerase chain reaction (PCR) (10−4) cells at 3 time points (day +11, day +24, and week +16 after chemotherapy). Three groups: low risk (LR; MRD− at both day +11 and day +24), intermediate risk (IR; neither LR nor HR), and high risk (HR; MRD+ until week +16) were defined and had distinct disease-free survival (DFS) and OS (Figure 4). MRD is also prognostic in the R/R setting. Hay et al23 studied patients with R/R B-ALL enrolled in a phase 1/2 clinical trial evaluating lymphodepleting chemotherapy followed by CAR T cells. EFS and OS were significantly better in the patients who achieved a MRD− CR compared with those in an MRD+ CR (median EFS, 7.6 vs 0.8 months, P < .0001; median OS, 20.0 vs 5.0 months, P = .014).
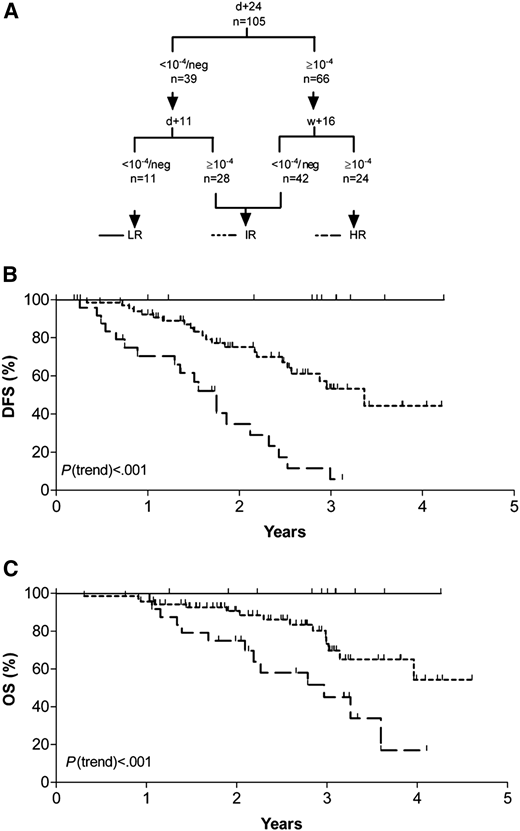
MRD-based risk groups. (A) Categorization schematic representation according to combined MRD results of day 11, day 24, and week 16. (B) Probability of disease-free survival (DFS). (C) Probability of overall survival (OS). LR, low-risk group; IR, intermediate-risk group; HR, high-risk group. Reproduced from Brüggemann et al18 with permission.
MRD-based risk groups. (A) Categorization schematic representation according to combined MRD results of day 11, day 24, and week 16. (B) Probability of disease-free survival (DFS). (C) Probability of overall survival (OS). LR, low-risk group; IR, intermediate-risk group; HR, high-risk group. Reproduced from Brüggemann et al18 with permission.
Although MRD is clearly important prognostically, our clinical application of MRD results is evolving. Patients who are MRD+ are at high risk for relapse. Historically, these patients have undergone transplantation in CR1 where transplantation has salvaged some (4-year DFS of 33% with transplant).24 Intensifying chemotherapy alone has altered the timing of relapse but has not improved the poor outcomes.25 MRD+ patients serve as an appropriate group in which new therapies can be tested (eg, blinatumomab and CAR T cells). Given the recent US Food and Drug Administration (FDA) approval of blinatumomab for MRD+ ALL, we generally treat MRD+ patients with blinatumomab followed by HCT in CR1, even if the patient achieves complete molecular remission (CMR) with blinatumomab. Outcomes with AHSCT are better for patients in CMR at the time of transplant.
With drugs such as blinatumomab, the rate of achieving molecular remission is increasing and a subset of patients experience long-term survival without transplant.26 In a patient such as the one presented here, CMR may help guide the decision not to transplant. Patients with comorbidities placing them at high risk for transplant-related mortality who are in CMR may not require HCT. For Ph+ ALL, HCT in CR1 has traditionally been recommended (discussed in the “Ph+ ALL” chapter). However, outcomes with second- and third-generation TKIs in combination with chemotherapy without HCT have been encouraging. Short et al examined the outcomes of patients with Ph+ ALL (n = 85) who received hyper CVAD (cyclophosphamide, vincristine, adriamycin, dexamethasone) and a TKI and no HCT.27 MRD measured by quantitative PCR for BCR-ABL at 3 months predicted both relapse-free survival (RFS) and OS; patients with CMR at 3 months had favorable long-term outcomes even without transplant. Similarly, early T-cell precursor (ETP) ALL has been typically associated with a high rate of refractory disease and poor prognosis. Historically, these patients were also transplanted in CR1. In COG AALL00434, Wood et al demonstrated that for those patients achieving end-induction CMR, the rates of 5-year EFS and OS were not inferior for patients with the ETP or near ETP subtype, suggesting that transplant decisions should be more heavily based on MRD than on subtype.28
Methods to measure MRD
Although MRD remains a critical prognostic marker, there are multiple methodologies to measure MRD and with different levels of sensitivity. It can be measured using multiparameter immunophenotypic evaluation of aberrant protein expression (multicolor 8-10 color flow), clone-specific PCR amplification of immunoglobulin and T-cell receptor gene rearrangements, or next -generation sequencing (Clone Seq; Adaptive) (Table 1). The level of sensitivity and specific time points used to make clinical decisions is highly protocol dependent. The FDA granted approval last year for the first assay to use next-generation sequencing to test for residual cancer cells following treatment in ALL. Clone Seq offers great sensitivity by sequencing across both immunoglobulin receptor gene sequences as well as frequently translocated regions of the genome (1 cell/106).29 Our institutions send MRD testing for multicolor flow cytometry to a centralized laboratory at the University of Washington in Seattle. We typically send testing at diagnosis (to assess the diagnostic clone), end induction, and 3 months into therapy, and then make subsequent decisions depending on the results.
Methods to measure MRD in ALL and sensitivities of the various approaches
| Method | Sensitivity |
|---|---|
| Next-generation sequencing | 10−6 |
| Flow cytometry | 10−4 |
| PCR for specific genes (ie, bcr-abl) | 10−5 |
| PCR immunoglobulin/T-cell receptor genes | 10−4 to 10−5 |
| Method | Sensitivity |
|---|---|
| Next-generation sequencing | 10−6 |
| Flow cytometry | 10−4 |
| PCR for specific genes (ie, bcr-abl) | 10−5 |
| PCR immunoglobulin/T-cell receptor genes | 10−4 to 10−5 |
Novel agents in different clinical settings
Rituximab and its role in MRD
CD20 positivity (defined as expression on ≥20% blasts) occurs in ∼40% of patients with B-ALL and has been associated with a worse prognosis. CD20 is upregulated with chemotherapy, making it an even more attractive therapeutic target.30 The monoclonal anti-CD20 antibody rituximab is recommended in the treatment of newly diagnosed CD20+, Ph− B-ALL patients <60 years of age based on a phase 3 trial demonstrating improvement in estimated 2-year EFS from 52% to 65% in patients receiving rituximab in addition to chemotherapy.31 Previous phase 2 trials by GMALL32 and MD Anderson33 demonstrated improved EFS and OS with the addition of rituximab compared with historical controls. The improvement in outcome with rituximab may be related to the effect of the antibody on MRD. In GMALL 7 in 2003, the rituximab arm achieved molecular CR (MRD < 10−4) at day 21 in 60% of patients compared with 19% in the control arm and at week 16 in 89% compared with 57% of patients.32 Current studies are evaluating the effectiveness of newer anti-CD20 antibodies such as obinituzumab.
Blinatumomab treatment of MRD
The prognosis of patients with postinduction MRD has improved significantly with the use of blinatumomab. It is a bispecific T-cell antibody with an anti-CD3 arm that engages T cells and an anti-CD19 arm that engages B lymphoblasts, leading to activation and proliferation of the cytotoxic T cells and lysis and apoptosis of the blasts.34 CD19 is almost uniformly expressed on all B-ALLs. Although blinatumomab was initially FDA approved for the treatment of R/R ALL, evidence of its activity was initially demonstrated in a pilot study in patients with MRD+ ALL.35 A subsequent large confirmatory study in MRD+ B-ALL patients demonstrated a 78% rate of CMR and improved RFS and OS in those patients treated with blinatumomab alone who achieved a CMR vs those who did not.36 Blinatumomab is FDA approved for the treatment of adult and pediatric patients with B-ALL in CR1 or CR2 with MRD ≥ 0.1%. Long-term remission has occurred in a subset of MRD+ patients who achieved CMR with blinatumomab and did not proceed to transplant. Twenty-five percent of patients without transplant or chemotherapy after blinatumomab remain in CR, as compared with 49% with HCT (median follow-up 24 months).36 Development of biomarkers to determine which patients do not require transplant would be a great advancement. However, until we can better predict this, suitable candidates who achieve CMR should undergo transplantation.
Blinatumomab for R/R B-ALL
Historically, the 5-year OS of adults in first relapse has only been 10%.37 Blinatumomab was first FDA approved for R/R B-ALL. Because of the drug’s short half-life, it is administered via continuous infusion. Because of its mechanism of action and the risk of cytokine release syndrome, patients must be admitted for the first 9 days; if the drug is interrupted for >4 hours (once outpatient), patients should be readmitted. In a phase 2 multicenter trial, 189 R/R Ph− pre-B-ALL patients were treated with blinatumomab.38 Patients with bone marrow blasts >50% received pretreatment with dexamethasone for up to 5 days to reduce the incidence of severe cytokine release syndrome. The median patient age was 39 years (range, 18-79 years), 39% of patients were in salvage 2 or higher, and 34% had received a prior HCT. Blinatumomab was well tolerated with the most common grade 3/ 4 adverse events being neutropenia, febrile neutropenia, and anemia.38 Three patients (2%) developed cytokine release syndrome and 52% of patients had neurologic events of which 76% were grade 1 or 2. This is indicative of blinatumomab’s immune-mediated mechanisms of action. The response rate of 43% CR/CR with incomplete count recovery (CRi), is encouraging given the heavily pretreated population. Eighty-two percent of responding patients achieved a CMR and 40% went on to transplant.38 The median RFS and OS were 5.9 months and 6.1 months, respectively. A subsequent randomized trial of blinatumomab vs standard of care chemotherapy in patients with R/R ALL confirmed a superior rate of CR/CRi (44% vs 25%) and median OS (7.7 months vs 4.0 months) for patients treated with blinatumomab.39
Blinatumomab as upfront therapy in elderly ALL patients
Current trials are examining blinatumomab as part of upfront treatment, including E1910 in which our patient participated. This trial compares the OS of patients with Ph− B-ALL who are MRD− following induction and intensification receiving blinatumomab with chemotherapy to those receiving chemotherapy alone. Trial S1318 evaluates blinatumomab as a single agent in newly diagnosed elderly ALL patients (≥65 years of age). Ph− B-ALL patients receive blinatumomab as induction (1-2 cycles) and postremission therapy (3 cycles) followed by prednisone, vincristine, 6-mercaptopurine, and methotrexate (POMP) maintenance. Preliminary results in 29 eligible patients, median age 75 years (range, 66-84 years), were presented at the 2018 ASH annual meeting.40 The most common grade 3-4 nonhematologic toxicities were hyperglycemia (14%), dyspnea (10%), and lung infection (7%). One patient developed grade 3 cytokine release syndrome and 1 patient developed grade 3 neurotoxicity. No patients died during the first 28 days of treatment. The overall response rate (CR/ CRi) was 66% (all CRs). Thirteen of 19 responders had MRD data available posttreatment and 12 (92%) were MRD− by multicolor flow cytometry. At 1 year, OS is 65% (95% CI, 43% to 80%) and DFS 56% (95% CI, 31% to 75%). Further follow-up will determine the durability of these responses and whether this approach has merit in the elderly, where many patients are poor candidates for intensive chemotherapy and historically the OS is only 10%. A subsequent currently accruing trial (A041703) builds on this approach with the sequential administration of inotuzumab for 1 to 2 cycles followed by blinatumomab for 4 to 5 cycles in newly diagnosed elderly patients (≥60 years of age) with B-ALL.
Inotuzumab in R/R ALL
CD22 is also expressed in the vast majority of B-ALLs. Inotuzumab ozogamicin is an anti-CD22 antibody attached through a linker to the cytotoxic agent calicheamicin. It is now FDA approved for patients with R/R B-ALL. An initial trial in lymphoma informed dosing and toxicities. A Phase 2 trial in R/R B-ALL performed at MD Anderson demonstrated promising results41 in heavily pretreated patients: 74% of patients in at least salvage 2 and 42% with poor risk cytogenetics. CR/CRi rate (18% CR, 39% CRi) was 57% with 63% of responders achieving CMR. Many patients did not recover platelets between cycles but still achieved CMR. Aside from common grade 3/4 myelosuppression, the drug was otherwise well tolerated. Drug-related fever occurred in 9 patients.41 Weekly fractionated dosing provides a more favorable toxicity profile than every 3 to 4 week dosing. Elevations in bilirubin and transaminases were generally grade 1 and grade 2 (55% and 24%, respectively); however, of 22 patients proceeding to HCT, 5 developed hepatic veno-occlusive disease (VOD). Four of these 5 patients received a transplant preparative regimen of clofarabine/thiotepa and clofarabine has known hepatic toxicity. Subsequent trials demonstrated a lower incidence of VOD.42 A randomized phase 3 trial of inotuzumab vs standard chemotherapy in patients with R/R B-ALL due for salvage 1 or salvage 2 demonstrated significantly higher CR/CRi rates in patients receiving inotuzumab (80.7% vs 29.4%).43 The rate of CMR among responders was also significantly higher with inotuzumab (78.4% vs 28.1%). The rate of VOD following HCT was 11%. In multivariable analysis, dual-alkylating condition therapy was the only significant covariate for VOD (P = .0039).43 OS favored inotuzumab (7.7 vs 6.7 months. P = .04) and the curves have more clearly separated with longer follow up (2-year OS rates of 22.8% vs 10.0%, P = .01).44 More patients receiving inotuzumab proceeded to HCT and patients who went directly on to HCT after attaining remission had favorable outcomes (median OS post-HCT not yet reached and 2-year OS probability of 51%).45
For patients with R/R B-ALL who require salvage therapy but will be proceeding to AHSCT, we favor blinatumomab over inotuzumab given the potential risk of VOD with inotuzumab. However, if a patient has significant tumor burden (including extramedullary disease) or has central nervous system disease, we recommend inotuzumab because response rate does not correlate with disease burden (bone marrow blast count).42 Blinatumomab’s response rate is lower in patients with high tumor burden.38 In terms of central nervous system disease, there is theoretical concern about potential toxicity with the combination of intrathecal therapy and blinatumomab. In treating a patient with inotuzumab, we administer as few cycles as possible prior to proceeding to AHSCT and leave time (2-3 weeks) between the last dose of inotuzumab and transplant (with the hope of reducing the risk of VOD), although other therapy may need to be given in the interim to keep the patient in remission.
Inotuzumab in the upfront setting
Given the encouraging results in the relapsed setting, inotuzumab has now been incorporated in the upfront setting in clinical trials. A current trial in AYAs (A041501) with newly diagnosed CD22+ B-ALL patients compares EFS using the C10403 regimen46 with and without inotuzumab. In elderly patients with newly diagnosed ALL, a phase 2 trial of mini-hyper CVAD plus inotuzumab, performed at MD Anderson treated 52 patients with a median age of 68 years (range, 64-72 years).47 Of previously untreated patients, 98% achieved CR/CRi and 78% achieved CMR at the time of morphologic response. Estimated 2 year PFS and OS were 59% and 66%, respectively. The most frequent grade 3/4 adverse events were prolonged thrombocytopenia (81%), infections during induction (52%) and consolidation chemotherapy (69%), hyperglycemia, hypokalemia, increased transaminases (19%), hyperbilirubinemia (17%), and hemorrhage. The incidence of VOD was 8%. Twelve percent of patients died of adverse events deemed treatment related. Given these encouraging results, an intergroup phase 3 trial of mini-hyper CVD (cyclophosphamide, vincristine, dexamethasone) plus or minus inotuzumab is being planned in this population.
Bcl-2 inhibition in R/R disease
Although much of the recent focus of treatment has been on immune-based therapy, other agents have also demonstrated encouraging results. The bcl-2 inhibitor venetoclax was recently FDA approved in combination with low-dose chemotherapy for elderly patients with newly diagnosed AML. Bcl-2 family proteins are key regulators of intrinsic apoptosis. Alford et al detected high levels of bcl-2 and high sensitivity to bcl-2 inhibition by the BH3 mimetic compounds, in pediatric ALL blasts, suggesting that bcl-2 inhibition may be an effective treatment strategy in ALL.48 Peirs et al demonstrated that some immature TLX3 or HOXA primary T-ALL lines were also highly sensitive to bcl-2 inhibition,49 likely due to high bcl-2 expression in early T-cell precursors. Because ALL cells demonstrate high levels of bcl-2 and bcl-xl, targeting both molecules may be effective.50 A phase 1 clinical trial combines venetoclax with the bcl-xl inhibitor navitoclax in relapsed/ refractory ALL and lymphoblastic lymphoma patients ≥4 years of age.51 Patients may also receive chemotherapy on day 9 onwards with peg-asparaginase, vincristine, and steroids. Patients were heavily pretreated with 2 patients having received 6 and 8 prior lines of treatment. There were no dose-limiting toxicities at 400 mg orally of venetoclax and 25 mg orally of navitoclax daily. Of 9 evaluable patients, 7 of whom received chemotherapy, 5 achieved CR/CRi. The most common serious adverse event related to venetoclax and navitoclax was febrile neutropenia (2 patients). Future strategies may include the mcl-1–specific inhibitor which has also demonstrated synergy with venetoclax.52
Conclusion
The patient in our case study completed chemotherapy, blinatumomab, and maintenance therapy on E1910 and remains in a CR 4 years after diagnosis. He has had persistent hypogammaglobulinemia (possibly related to blinatumomab) but no infections. We are learning more about the molecular heterogeneity of ALL, which will lead to individualization of management. Combinations of chemotherapy with molecularly targeted therapy and/or antibody-based therapy will likely move upfront in the near future and further improve outcomes. MRD testing, particularly using highly sensitive techniques, will help better tailor therapy, especially in elderly patients and in patients for whom HCT is considered.
Correspondence
Anjali S. Advani, Cleveland Clinic Foundation, 10201 Carnegie Ave, Desk CA60, Cleveland, OH 44195; e-mail: advania@ccf.org.
