
Abstract
Proper diagnostic distinction of bone marrow failure syndromes can often be challenging. In particular, for older patients with idiopathic aplastic anemia (AA), differential diagnosis includes myelodysplastic syndrome (MDS), which can atypically present in a hypocellular form. In addition to blasts and overt dysplasia, the presence of chromosomal abnormalities and a spectrum of somatic mutations may be revealing. Both clonal cytogenetic aberrations and somatic mutations most typically correspond to a clonal myelodysplasia, but clonal somatic mutations have also recently been found in AA. True driver myeloid mutations are uncommon in AA. Marrow hypocellularity in AA and occasionally in MDS patients points toward a similar immune mechanism responsible for deficient blood cell production and indicates that cytopenias in early hypocellular MDS might be treated with immunosuppressive modalities. Primary hypocellular MDS has to be distinguished from post-AA secondary MDS, most commonly associated with del7/7q. Post-AA MDS evolves at the rate of about 10% in 10 years, but recent observations suggest that widespread use of eltrombopag may influence the risk of progression to MDS. This complication likely represents a clonal escape, with founder hits occurring early on in the course of AA. A similar mechanism operates in the evolution of paroxysmal nocturnal hemoglobinuria (PNH) in AA patients, but PNH clones are rarely encountered in primary MDS.
Learning Objectives
Understand aspects of clonality, clonal hierarchy, and dynamics in current concepts of bone marrow failure and hematopoiesis in young and older individuals
Establish pathogenetic relationship between AA and hypocellular MDS
Understand the diagnostic differences between AA and hypocellular myelodysplasia in the genomic era
Acquire the ability to infer clinical information and improve skills for interpreting deep next-generation sequencing in bone marrow failure syndromes
Introduction
Despite the apparent simplicity of the pathomorphologic picture, the diagnosis of acquired aplastic anemia (AA) may be problematic in real case scenarios. During childhood, congenital bone marrow failure syndromes account for the majority of diagnostic challenges,1-3 whereas in adults and the elderly, the differential diagnosis of pancytopenia with empty bone marrow includes myeloid neoplasia, in particular, hypocellular myelodysplastic syndrome (MDS) and the rare antiquated entity of aleukemic leukemia. Clonal evolution and the interaction of aberrant clones with immune response directed against hematopoietic stem and progenitor cells are the key pathogenetic aspects of AA, secondary MDS evolved from AA, primary MDS, and its special form, hypocellular MDS (Figure 1). In recent years, our knowledge of the molecular pathogenesis of AA has been greatly enriched by systematic application of next-generation sequencing (NGS). Deep NGS panel assays for detection of somatic mutations are now routinely available and provide prognostic as well as diagnostic clues helpful in distinguishing AA from congenital bone marrow failure syndromes in younger patients and hypocellular MDS in older patients. Both somatic and germ line alterations can be integrated into the current models of AA pathogenesis, and investigations have begun to explore the usefulness of these novel tools to manage AA. However, higher throughput, higher sensitivity, and a wider application of clinical sequencing for the diagnosis of somatic mutations have also resulted in new challenges of clonality issues, such as the presence of clonal hematopoiesis of indeterminate potential (CHIP) and the detection of numerous variants of unclear significance of germ line and somatic origin.4
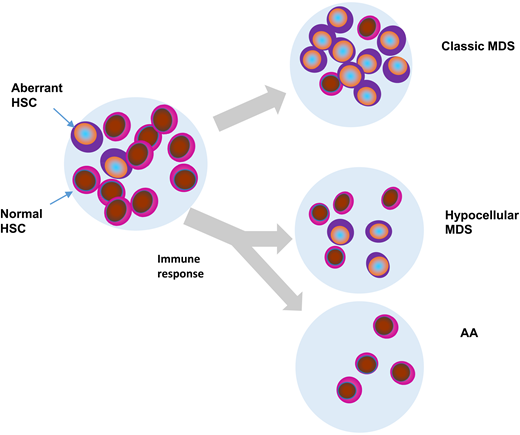
Hypothetical fates of aberrant myeloid clones in AA, typical MDS, and hypocellular MDS. HSC, hematopoietic stem cells.
Hypothetical fates of aberrant myeloid clones in AA, typical MDS, and hypocellular MDS. HSC, hematopoietic stem cells.
Clinical overlap between AA and MDS
There are several pathogenetic interjections between AA and MDS. Some of them have been revealed by systematic application of NGS to study AA and MDS (Figure 1). They include coincidental presence of CHIP in AA, fluctuating clonal hematopoiesis vs otherwise well-described evolution of MDS from AA, diagnostic overlap between AA and hypocellular MDS, and conceptual similarities between paroxysmal nocturnal hemoglobinuria (PNH) and MDS.
AA vs hypocellular MDS
The bimodal demographics of idiopathic AA with a second incidence peak occurring in older individuals clearly predisposes them to diagnostic challenges in distinguishing their disease from MDS. Approximately 10% to 20% of MDS cases can occur in the context of empty bone marrow, a problem amplified by the decline in cellularity inherent to aging. Currently, the most accepted notion is that cellularity of ≤25% would be defining of hypocellular MDS based on the cutoff value of 20% to 30% originally used for AA. However, this definition does not take into account age-related cellularity loss.5 In addition, assessments of cellularity are subjective and may be impaired by sampling errors, patchy marrow appearance, and observer bias. Nevertheless, a hypocellular subtype of MDS does indeed exist (known as hypoplastic or hypocellular MDS) as a bewildering differential diagnostic consideration, especially in older individuals.6,7
US hematologists link the diagnosis of AA to the presence of a normal karyotype, whereas a finding of abnormal cytogenetics in a significant fraction of metaphases is rather more compatible with the diagnosis of MDS. Exceptions to this rule include some benign random aberrations and/or clonal abnormalities, including the loss of the X or Y chromosome in elderly females or males, respectively.8,9 Whereas not strictly pathogenic, detection of such abnormalities suggests at minimum the presence of (oligo)clonal hematopoiesis. However, the criterion of strictly normal karyotypes in AA is somewhat controversial; in some studies, the presence of certain karyotypic abnormalities (eg, trisomy 8) were considered compatible with the diagnosis of AA. Nevertheless, most hematologists believe that cytogenetics must be normal in typical AA.10 The presence of myeloid dysplasia and macrocytosis (without the presence of PNH clones) is suggestive of MDS in older individuals. Abnormal cytogenetics such as aneuploidy and structural abnormalities confirm this prediction. Because abnormal cytogenetics are encountered in only ∼50% of patients with MDS, a normal karyotype does not simply exclude the diagnosis. Clearly, hyper- or normocellular marrow helps in diagnostic appropriation, but a fraction of patients may in fact have a hypocellular form of MDS. The presence of PNH may also be helpful, because in some studies, tiny PNH clones were encountered in 30% to 60% of patients with idiopathic AA.11 PNH is nearly absent in normal primary MDS (∼2%); thus, the occasional presence of PNH clones in the setting of hypocellular MDS may point toward the diagnosis of AA or toward the development of secondary MDS from primary AA. Although progression rate to MDS may be as high as 20% in 20 years,12,13 it is most commonly associated with increasing marrow hypercellularity. Similarly, in PNH or AA syndrome, hypocellularity is replaced by erythroid hyperplasia. Tables 1 and 2 summarize some diagnostic principles and compare some key morphologic differences. Certain histologic and cytologic parameters can potentially be useful in differentiating between AA and hypocellular MDS. Although both entities may present with erythroid dysplasia and mild to moderate myeloid dysplasia, other features such as dysmegakaryopoiesis and severe dysgranulopoiesis are considered highly specific for primary MDS, including hypocellular MDS.5 Ringed sideroblasts, bone marrow fibrosis, and clusters of CD34+ cells are also more common in MDS. Of special note is the incompatibility of blasts with the diagnosis of AA. Both AA and hypoplastic MDS may respond to immunosuppressive therapy, whereas classic MDS is less likely to respond, which supports the case for a shared immune-mediated pathology in hypocellular MDS and AA. Overall, the detection rate of chromosomal abnormalities, and thereby better distinction from AA, was improved with a more precise single nucleotide polymorphism (SNP) karyotyping, which if combined with metaphase cytogenetics yields cytogenetic changes in 54% of hypoplastic MDS cases compared with 19% of AA cases.14,15 The remaining patients have to be carefully evaluated, because dysplastic changes, when limited to erythroid series in particular, are not diagnostic for MDS, but any elevation of blasts should point to a diagnosis of MDS irrespective of cytogenetics.16 Diagnosis of hypocellular MDS should not be invoked in patients who received hypomethylating agents or other forms of chemotherapy.
Contrasting features of hypocellular MDS vs AA
| Age | Cytogenetics | PNH clone | Blasts | Dysplasia | Macrocytosis | Somatic mutations | |
|---|---|---|---|---|---|---|---|
| AA | Bimodal | Normal | 20%-30% | Absent | Erythroid only | With PNH | Absent, low variant allelic frequency, absence of splicing mutations |
| Hypocellular MDS | Older | 50% abnormal | Absent | Normal increased | Trilineage or bilineage | Prevalent | Canonical MDS hit, including spliceosomal mutations |
| Age | Cytogenetics | PNH clone | Blasts | Dysplasia | Macrocytosis | Somatic mutations | |
|---|---|---|---|---|---|---|---|
| AA | Bimodal | Normal | 20%-30% | Absent | Erythroid only | With PNH | Absent, low variant allelic frequency, absence of splicing mutations |
| Hypocellular MDS | Older | 50% abnormal | Absent | Normal increased | Trilineage or bilineage | Prevalent | Canonical MDS hit, including spliceosomal mutations |
Morphologic, prognostic, and immunosuppression therapy response distribution
| Characteristic | AA | Post-AA MDS | Hypoplastic MDS | MDS |
|---|---|---|---|---|
| Marrow cellularity | Decreased | Mostly Increased | Decreased | Normal/ increased |
| Abnormal neutrophils | − | possible | + | + |
| Abnormal megakaryocytes | − | − | + | + |
| Blasts | Absent | Increased | +/− | Often increased |
| Megakaryocytes | Absent/decreased | Absent | + | + |
| Fibrosis | − | − | Occasional | Occasional |
| Progression | ∼10% | + | >25% | >25% |
| Response to IST | ++ | − | + | − |
| PNH defect | ∼30% | Possible | Rare | Absent |
| T-cell activation | + | + | + | +/− |
| LGL | − | − | + | + |
| Characteristic | AA | Post-AA MDS | Hypoplastic MDS | MDS |
|---|---|---|---|---|
| Marrow cellularity | Decreased | Mostly Increased | Decreased | Normal/ increased |
| Abnormal neutrophils | − | possible | + | + |
| Abnormal megakaryocytes | − | − | + | + |
| Blasts | Absent | Increased | +/− | Often increased |
| Megakaryocytes | Absent/decreased | Absent | + | + |
| Fibrosis | − | − | Occasional | Occasional |
| Progression | ∼10% | + | >25% | >25% |
| Response to IST | ++ | − | + | − |
| PNH defect | ∼30% | Possible | Rare | Absent |
| T-cell activation | + | + | + | +/− |
| LGL | − | − | + | + |
IST, immunosuppressive therapy; LGL, large granular lymphocyte leukemia; +, presence and/or response; −, negative response or absence.
In recent studies, hypocellular MDS has been contrasted to normocellular or hypercellular classic MDS and AA in terms of somatic mutational pattern.17 Not surprisingly, a mutational pattern of hypocellular MDS seems to overlap with that of classic MDS, except for the relative absence of spliceosomal mutations often associated with either ring sideroblasts (SF3B1) or with myeloproliferative features (SRSF2, ZRSR2, U2AF1), which are the typical drivers of mutations in classic MDS.17,18 Recently, clonal myeloid mutations have also been found in a large proportion of patients with AA.12,19,20 Comparisons of mutational spectra to hypocellular MDS showed a similar pattern, but BCOR/BCORL mutations seemed to be encountered frequently, whereas TET2 mutations were less common in AA.5,20 Importantly, the somatic pattern in hypocellular MDS was rather distinct from that observed in secondary MDS evolved from AA, whereby mutations in RUNX1, SETBP1, and ASXL1 and foremost del7/7q were more prevalent in post-AA MDS (Figure 2).18
![Differential distribution of somatic mutations in AA, MDS (hypoplastic [Hypo] and hyperplastic [hyper]), and PNH. The red bars depict the percentage of patients affected by a given somatic mutation. Proportion of patients with PNH clones (PIGA mutation) is indicated in blue. Adapted from Negoro et al.18 IST, immunosuppressive therapy; Normo, normocellular; sMDS, secondary myelodysplastic syndrome: sMDS refers to MDS after AA.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2019/1/10.1182_hematology.2019000019/5/m_hem2019000019cf2.png?Expires=1763474035&Signature=rK278iuHErsHrdeaqGETuZcHRlxJXCSwoCRTHqpUfH9j1wJrGQquGrlhN~ytLbD0pRJQZVOW4GsZYxrmA8khtv3ZhWOLQCxcMEdcqv7zhgmFheyPGHXTDVWhks7MlpvUlzE2XnvJ5NkVf3B4pR8bexdRvVWRA-DNp2VxD9wQPXMMzZMO7cgANDNkaQNQVBhLXON5B9sh1h6oJ0plqngUVMaM3hVBAjOmKuDDD2TctpJVI9ESyKNoz5Cmz48gnD9FSMyYuhi2zwi2rowNLK64OpA~ENpnBKcYETytG0MSBoebTJsi13e5tjfeua28iZGVGe~68ofoHoX1WvKffhvbog__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Differential distribution of somatic mutations in AA, MDS (hypoplastic [Hypo] and hyperplastic [hyper]), and PNH. The red bars depict the percentage of patients affected by a given somatic mutation. Proportion of patients with PNH clones (PIGA mutation) is indicated in blue. Adapted from Negoro et al.18 IST, immunosuppressive therapy; Normo, normocellular; sMDS, secondary myelodysplastic syndrome: sMDS refers to MDS after AA.
Differential distribution of somatic mutations in AA, MDS (hypoplastic [Hypo] and hyperplastic [hyper]), and PNH. The red bars depict the percentage of patients affected by a given somatic mutation. Proportion of patients with PNH clones (PIGA mutation) is indicated in blue. Adapted from Negoro et al.18 IST, immunosuppressive therapy; Normo, normocellular; sMDS, secondary myelodysplastic syndrome: sMDS refers to MDS after AA.
In a recent study, clinical morphologic, cytogenetic, and mutational parameters were used to devise a diagnostic scoring system that allows for a formalized distinction between AA and hypocellular MDS (Table 3). The higher the score, the more likely the diagnosis of hypocellular MDS is, and scores ≥2 are highly suggestive of this disease.
Proposed schema for differentiating bone marrow failure syndromes hypocellular MDS and aplastic anemia
| Cytologic/histologic variables | Score |
|---|---|
| Blasts ≥5% | 2 |
| Blasts 2%-4% | 1 |
| Fibrosis grade 2-3 | 1 |
| Dysmegakaryopoiesis | 1 |
| Ringed sideroblasts ≥15% | 2 |
| Ringed sideroblasts 5%-14% | 1 |
| Severe dysgranulopoiesis | 1 |
| Abnormal cytogenetics | 2 |
| MDS corresponding mutational pattern | 1 |
| Cytologic/histologic variables | Score |
|---|---|
| Blasts ≥5% | 2 |
| Blasts 2%-4% | 1 |
| Fibrosis grade 2-3 | 1 |
| Dysmegakaryopoiesis | 1 |
| Ringed sideroblasts ≥15% | 2 |
| Ringed sideroblasts 5%-14% | 1 |
| Severe dysgranulopoiesis | 1 |
| Abnormal cytogenetics | 2 |
| MDS corresponding mutational pattern | 1 |
Scores ≥2 are more specific for hypocellular MDS compared with AA. Adopted from Bono et al.5
Recent studies comparing outcomes in patients with MDS with outcomes for patients with AA and/or classic MDS showed a worse overall survival in patients with classic MDS compared with those with hypocellular marrow and an inverse relationship with acute myeloid leukemia progression, whereas AA patients showed a better prognosis. The aforementioned scoring system devised for distinguishing MDS and hypocellular MDS (Table 3) also correlated with survival; patients with a high score showed outcomes similar to those of patients with classic MDS.5
In terms of clinical management, the presence of hypocellular marrow may be important in the setting of low-risk disease. Original observation of responsiveness of MDS to immunosuppression in a few isolated patients and small series has been followed by several trials. In one of these trials from the National Institutes of Health, the response rate in selected refractory patients approached 35%. Among these patients, higher responses were observed in refractory anemia and in hypocellular MDS (48%).21,22 Responses have also been reported for treatment with cyclosporin A in 6 of 8 patients with hypocellular MDS. However, other groups were able to only partially reproduce these results. In a recent meta-analysis, responses (complete and partial combined) in the range of about 17% of patients have been reported, with hypocellular marrow serving as a positive predictive factor for responsiveness to Anti-thymocyte globulin.23 Other factors, such as the presence of HLA-DR15 and younger age (reminiscent of AA), also point toward the rational application of immunosuppression in this subtype of MDS.21,23 In general, abnormal highly clonal cytogenetics, the presence of blasts, or the presence of high-risk mutations argue against the use of immunosuppressive therapy. The relative effectiveness of this modality in hypocellular MDS and AA points toward common pathogenetic mechanisms (Table 2). In contrast to immunosuppression, a few systematic studies of MDS predict response to hypomethylating agents, but in general, empty marrow is not a positive predictive factor.24 The presence of hypocellular marrow, even in the setting of otherwise overt MDS, suggests that therapy with immunosuppression, including ATG, may be effective.23
Post-AA secondary MDS vs hypocellular MDS
Secondary MDS can evolve during the course of idiopathic AA (Figure 3; Table 2). Most commonly, post-AA MDS is characterized by the presence of monosomy 7 or del7q; other cytogenetic changes are less common.18 SNP karyotyping can occasionally detect uniparental disomy 7q, or other somatic uniparental disomies that would be not detectable by metaphase cytogenetics.14 The progression rate has been estimated to be 10% to 20%.13,18 Recently, concerns have been raised regarding whether the increasing use of eltrombopag will lead to a higher rate or possibly an earlier onset of clonal transformation.25
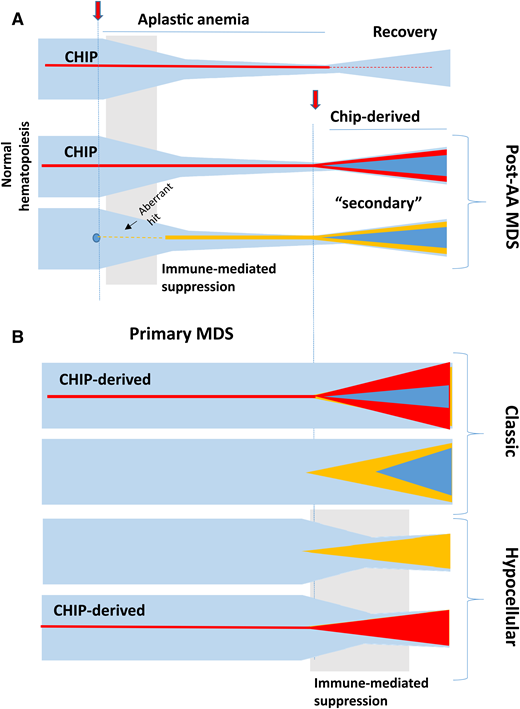
Clonal evolutionary chart of bone marrow failure syndromes over time for AA and primary MDS. Normal hematopoiesis would, if not influenced by a disease pathology, be expected to remain relatively stable with only a slow age-related decline. (A) CHIP, random or pathogenic aberrant myeloid clones, or idiopathic triggers can induce an immune reaction against the hematopoietic stem and progenitor cell (HSPC) compartment leading to AA with contraction of the HSCP pool. Transient or persistent benign clonality may result from CHIP. Either CHIP or another de novo aberrant clone may be an origin of clonal progression (eg, by the acquisition of additional genetic hits resulting in post-AA secondary MDS). (B) Similarly, primary MDS including its classic (hypercellular or normocellular) or atypical (hypocellular) forms can evolve from a slow-driving preexisting CHIP clone or an aberrant de novo somatic hit. Hypocellularity may be a result of immune attack stemming from a tumor surveillance reaction similar to idiopathic AA.
Clonal evolutionary chart of bone marrow failure syndromes over time for AA and primary MDS. Normal hematopoiesis would, if not influenced by a disease pathology, be expected to remain relatively stable with only a slow age-related decline. (A) CHIP, random or pathogenic aberrant myeloid clones, or idiopathic triggers can induce an immune reaction against the hematopoietic stem and progenitor cell (HSPC) compartment leading to AA with contraction of the HSCP pool. Transient or persistent benign clonality may result from CHIP. Either CHIP or another de novo aberrant clone may be an origin of clonal progression (eg, by the acquisition of additional genetic hits resulting in post-AA secondary MDS). (B) Similarly, primary MDS including its classic (hypercellular or normocellular) or atypical (hypocellular) forms can evolve from a slow-driving preexisting CHIP clone or an aberrant de novo somatic hit. Hypocellularity may be a result of immune attack stemming from a tumor surveillance reaction similar to idiopathic AA.
Cytopenias with a protracted course that lack improvement or secondary recurrence are frequent signs of clonal progression. In fact, lack of response is likely a predisposing factor to clonal evolution.26 Change in the marrow cellularity, appearance of clonal cytogenetic abnormalities (chiefly −7/del7q18 ), overt dysplasia, or new clonal driver mutations commonly found in MDS herald the evolution of MDS. However, in some cases, in the setting of severe cytopenia, MDS evolution can be diagnosed despite persisting hypocellularity through the detection of cytogenetic abnormality or the presence of blasts. This would constitute a special form of hypocellular MDS, distinct from primary hypocellular MDS.
Sequencing studies have shown that in some patients, progression is associated with expansion of clones characterized by the presence of driver clonal abnormalities in RUNX1, SETBP1, or CBL for example (Figure 2).18 Indeed, founder mutations may have been present at the onset of AA,27 with many pathogenetic implications (discussed in the forthcoming paragraphs).28 It is possible that founder somatic mutations are followed by del7/7q. Analysis by allelic imbalance methods showed that, in some patients, initial mutations are followed by a deletion event, whereas in other patients, del7 is the primary lesion. Factors leading to the predilection to del7/7q remain unclear.29 It is possible that germ line predisposition (eg, GATA2 abnormalities), SAMD9/SAMD9L variants, or other genetic factors may play a role in the evolution of AA and/or in the propensity to progression.30,31 Somatic mutational spectra are shown in Figure 2.
Case 1
A 71-year-old man presented with lethargy, bruising, shortness of breath, and chest pain on exertion. Initial workup ordered by the primary care physician revealed pancytopenia with a white blood cell count of 2690/μL; hemoglobin, 6.5 g/dL; mean corpuscular volume (MCV), 109.1 fL; platelet count, 15 000/μL; reticulocytes, <1%; absolute neutrophil count, 990/μL; lactate dehydrogenase, 260 U/L; and haptoglobin 20 mg/dL. Repeat complete blood count showed persistent pancytopenia. The patient was referred to hematology with a presumptive diagnosis of MDS. Bone marrow biopsy revealed reduced cellularity (15%), 0% blasts, and a decreased myeloid to erythroid precursors (M:E ratio). No ringed sideroblasts were noted, granulopoiesis showed complete maturation, and erythropoiesis exhibited megaloblastic changes with mild dysplasia, including occasional binucleation. Megakaryocytes were absent. Cytogenetics did not recover any metaphases, but an NGS panel demonstrated the presence of DNMT3A fs. p.R882h mutation with a variant allele frequency (VAF) of 10%.
These initial findings suggest MDS with a typical seventh decade presentation, elevated MCV, dyserythropoiesis, DNMT3A mutation, and hypocellularity that could be explained by age compared with the hypocellular form of MDS. However, erythroid dysplasia is not sufficient for the diagnosis of MDS, with empty bone marrow and absent megakaryocytes and blasts. DNMT3A could represent coincidental CHIP, but clonal mutations can also be found in otherwise typical AA. Additional tests were performed: fluorescence in situ hybridization was negative for del7, del5q, del20q, and duplication of chromosome 8. SNP array karyotyping was negative for unbalanced translocations or areas of loss of heterozygosity (copy-neutral as well as deletions and duplications). Flow cytometry for PNH revealed the presence of a small glycosylphosphatidylinositol-negative clone size of 6% in granulocytes and 1% red blood cells.
Detection of a small PNH clone in the absence of any cytogenetic abnormalities reinforced the diagnosis of AA. The erythroid dysplasia, decreased M:E ratio, and increased MCV could be the result of PNH-related intravascular hemolysis. Blasts and megakaryocytes are typically absent in AA.
After discussion with the patient, a combination of antithymocyte globulin and solumedrol was administered in the hematology ward. The patient was discharged with cyclosporine 100 mg twice per day, magnesium supplementation, eltrombopag 75 mg twice per day, and a 4-week steroid taper.
Common pathogenesis
Various theories of AA pathogenesis explain the overlap between acquired AA and MDS, in particular hypocellular MDS (Figures 1 and 4). Clearly, some of the cytopenias presenting in early MDS, before a total infiltration of the marrow, in analogy to idiopathic AA, may be related to immune suppression with immunosuppressive therapies shown to have considerable efficacy.21,23,32 In AA, the triggers of immune attack are not well defined but may include autoantigens as a result of a breach of tolerance, viral antigens, autoantigens targeted through molecular mimicry, and epitopes from aberrant transformed cells (Figure 4A-B). The latter mechanisms would be compatible with cellular tumor surveillance reactions triggered by pre-existing aberrant myeloid clone (Figure 4C). In cases of AA, such responses may lead to the elimination of abnormal cells, but at the cost of collateral damage to normal hematopoietic stem and progenitor cells. However, if clonal evolution is not controlled or is only partially contained by the immune system, secondary MDS may evolve through acquisition of selective resistance and immune escape (Figure 4C). In hypocellular MDS, the triggers lead to suppression of hematopoiesis, but likely provide some control that prevents clonal expansion, whereas progression to full-blown MDS would be compatible with the secondary failure of tumor surveillance (Figure 3). Occasionally, patients with large granular lymphocyte leukemia and coinciding MDS support a similar notion, except it is likely that in large granular lymphocyte leukemia, the distribution of the antigenic triggers may be more limited to lineage-restricted progenitors.33
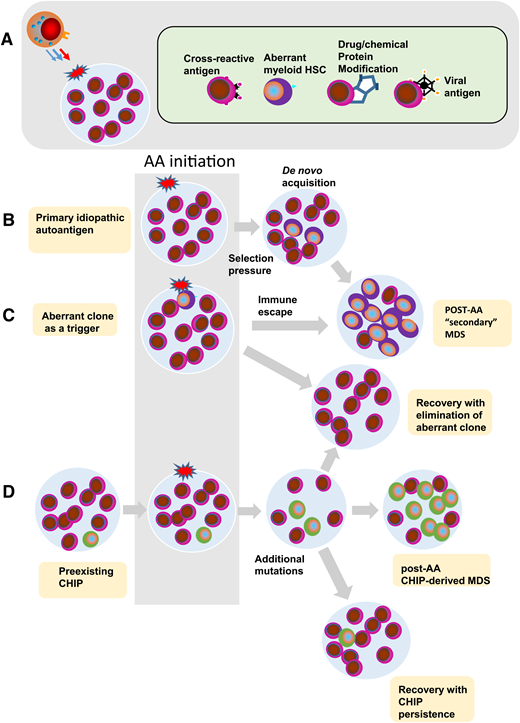
Pathogenetic theories of bone marrow failure as a result of interaction of the HSC compartment with the immune system. (A) Various triggers of a cytotoxic T-lymphocyte reaction in AA. (B) Immune attack against a hypothetical initiating event results in endogenous carcinogenesis and de novo acquisition of clonal events. Immune selection pressure can lead to further clonal evolution of secondary MDS after AA. (C) Theoretically, the aberrant clone itself may be a trigger leading to a similar pathogenic escape reaction and a similar outcome. It is also possible that either a nonspecific or an overshooting immune reaction leads to a contraction of the HSPC compartment as seen in AA or hypocellular MDS. Finally, ineffective tumor surveillance reaction fails to prevent clonal evolution analogous to classic or typical MDS. (D) Recent data demonstrate the presence of somatic mutant clones, consistent with CHIP, in patients with otherwise typical AA. In this scenario, immune response may further augment the oligoclonality by the removal of normal HSCs. Depending on the penetrance of somatic CHIP hits, they expand whereas normal cells are eliminated thus leading to hypocellular MDS. Alternatively, upon successful immunosuppression and recovery of the normal HSCs, the mutant clones are diluted out by expanded normal HSCs.
Pathogenetic theories of bone marrow failure as a result of interaction of the HSC compartment with the immune system. (A) Various triggers of a cytotoxic T-lymphocyte reaction in AA. (B) Immune attack against a hypothetical initiating event results in endogenous carcinogenesis and de novo acquisition of clonal events. Immune selection pressure can lead to further clonal evolution of secondary MDS after AA. (C) Theoretically, the aberrant clone itself may be a trigger leading to a similar pathogenic escape reaction and a similar outcome. It is also possible that either a nonspecific or an overshooting immune reaction leads to a contraction of the HSPC compartment as seen in AA or hypocellular MDS. Finally, ineffective tumor surveillance reaction fails to prevent clonal evolution analogous to classic or typical MDS. (D) Recent data demonstrate the presence of somatic mutant clones, consistent with CHIP, in patients with otherwise typical AA. In this scenario, immune response may further augment the oligoclonality by the removal of normal HSCs. Depending on the penetrance of somatic CHIP hits, they expand whereas normal cells are eliminated thus leading to hypocellular MDS. Alternatively, upon successful immunosuppression and recovery of the normal HSCs, the mutant clones are diluted out by expanded normal HSCs.
According to the competing, perhaps less popular theory, immune responses and inflammatory stress can result in DNA damage and acquisition of clonal cytogenetic defects and/or somatic mutations. Similarly, stress hematopoiesis may increase DNA damage through increased hematopoietic stem cell (HSC) turnover and replicative stress. In either case, aberrant clones would be secondary to the immune process (Figures 3 and 4D). Appearance of these effects in elderly patients argues that they are cumulative and have to be present over prolonged periods of time to result in disease.
In addition to these theories (primary and secondary evolution of MDS clones), the discovery of CHIP leads to other stipulations. For instance, CHIP may initiate clonal expansion in a proportion of MDS patients that may result in unopposed expansion or cause an immune reaction leading to hypocellularity secondary to cytotoxic immune mediation. In AA, some of the clonal changes may simply correspond to exposed CHIP in lieu of depletion (and oligoclonality) of normal HSCs (Figures 3 and 4D).
Case 2
A 48-year-old woman presented with complaints of lethargy over the last 2 months and recent concerns of increased lower extremity bruising. Complete blood count results showed pancytopenia, and she was referred to a hematologist for further evaluation. Pertinent laboratory results included white blood cell count, 0.44/μL; hemoglobin, 6.9 g/dL; MCV, 103.0 fL; platelet count, 16 000/μL; reticulocyte count, <0.4%; absolute neutrophil count, <30/μL; lactate dehydrogenase, 362 U/L; and haptoglobin, 30 mg/dL. The patient was receiving methimazole for hyperthyroidism. Considering her young age, she was referred to hematology with a presumptive diagnosis of AA. Bone marrow biopsy was hypocellular (18%), with trilineage hematopoiesis and proper maturation. Bone marrow aspirate showed 2% blasts and 35% erythroid precursors. Occasional small dysplastic megakaryocytes, macrocytic anemia with anisocytosis, polychromasia, and poikilocytosis, including ovalocytes, were seen. Ringed sideroblasts were within normal limits.
On the basis of her young age, history of methimazole previously implicated in AA, pancytopenia, and marrow hypocellularity, a diagnosis of AA was likely, but the presence of any blasts in AA is atypical. Moreover, megakaryocytes would be absent or significantly decreased and, if present at all, should have a normal appearance. The patient received methimazole for 2 years, which made acute presentation of drug-induced AA less likely.
Subsequently, PNH flow cytometry was obtained and was negative for GPI-deficient granulocytes typically found in AA. Ultimately, the results of cytogenetics arrived showing 45XX, −7 [4] and 46XX [16], thus making the diagnosis of hypocellular MDS more likely. Multiple somatic mutations were also found on the NGS panel, including ASXL1 p.Arg693* VAF, 30%; RUNX1 p.R207W VAF, 20%; SRSF2 p.Pro95His VAF, 36%.
Prognostic and diagnostic scoring for hypocellular MDS (Table 3) placed the patient at high risk for progression to acute myeloid leukemia with 4: 1 point for megakaryopoiesis, 2 points for abnormal cytogenetics, and 1 point for somatic mutations, especially when combined with a Revised International Prognostic Scoring System (IPSS-R) score of 6 (IPSS-R high category). The patient underwent a workup for HSC transplantation, and interval MDS FISH showed 80% cells with −7 further corroborating the diagnosis, and reaffirming the need for HSC transplantation. Upon completion of HSC evaluation, the patient was offered first-line matched related HSCs from a younger sister.
Conclusion
Analyzing the evolutionary progress can provide a fundamental understanding of the pathological processes and help guide diagnosis. The fate of the HSC compartment and aberrant hematopoietic clones correlates with disease stages and underlying intrinsic expansion drive vs counteracting immune responses (Figures 3-4).
New genomic technologies have advanced our understanding of the pathogenetic mechanisms of hypocellular MDS and its relationship to AA. Mutational profiling has improved the diagnostic specificity of identifying myeloid neoplasms and in differentiating between a possibly nonmalignant and a potentially malignant bone marrow failure syndrome. Although some genetic and clinical differences exist, potential applicability of immunosuppressive therapies remains a common feature of hypocellular MDS pointing toward immune pathogenetic aspects of hematopoietic failure in both AA and hypocellular MDS.
Correspondence
Jaroslaw P. Maciejewski, Department of Translational Hematology and Oncology Research, Lerner Research Institute, Cleveland Clinic, 9620 Carnegie Ave n building, NE6-314, Cleveland, OH 44106; e-mail: maciejj@ccf.org.
