

Abstract
Advances in the diagnosis and treatment of inherited bone marrow failure syndromes (IBMFS) have provided insight into the complexity of these diseases. The diseases are heterogeneous and characterized by developmental abnormalities, progressive marrow failure, and predisposition to cancer. A correct diagnosis allows for appropriate treatment, genetic counseling, and cancer surveillance. The common IBMFSs are Fanconi anemia, dyskeratosis congenita, and Diamond-Blackfan anemia. Hematopoietic cell transplantation (HCT) offers curative treatment of the hematologic complications of IBMFS. Because of the systemic nature of these diseases, transplant strategies are modified to decrease immediate and late toxicities. HCT from HLA-matched related or unrelated donors offers excellent survival for young patients in aplasia. Challenges include the treatment of adults with marrow aplasia, presentation with myeloid malignancy regardless of age, and early detection or treatment of cancer. In this article, I will describe our approach and evaluation of patients transplanted with IBMFS and review most frequent complications before and after transplant.
Learning Objectives
Examine the effect of age, disease and disease status at transplant, prior treatments including chronic transfusion and other disease-specific complications on transplant outcomes
An understanding of the consequences of transplant strategies as it pertains to the underlying disease and how these potentially add to the burden of morbidity and mortality associated with transplantation
Introduction
Although inherited bone marrow failure syndromes (IBMFS) are typically diagnosed in childhood or adolescence, an increasing number of patients may present to adult hematologists with atypical presentations. Careful consideration should be given to the patient’s history and physical findings, and family history needs to be investigated to detect other relatives with IBMFS, hematologic malignancies, solid tumors, and pulmonary or hepatic complications.1,2 Patients with IBMFS should undergo extensive clinical and laboratory evaluations before and after hematopoietic cell transplantation (HCT; Tables 1 and 2). A comprehensive review of systems involved in these syndromes was recently published by Alter in 2017.1 Clinical manifestations are heterogenous and have variable penetrance within affected members of the same family, and screening of family members is essential to exclude them as potential donors.1-4 (Figure 1) As IBMFS can impact organs other than the bone marrow, the care of these patients requires a multidisciplinary team. HCT is the treatment of choice for most patients with bone marrow failure and preferably with radiation-free reduced intensity conditioning (RIC) regimens.5-7 Bone marrow is the preferred graft in T-replete transplants, because peripheral blood is associated with higher risk for graft-versus-host disease (GVHD) and second malignancy.5,8,9
Pretransplant evaluation of patients with IBMFS
| • Complete family and patient’s history with careful evaluation of physical findings according to disease type. | ||
| • Multidisciplinary team: Visual, hearing, endocrine, nutritional, and neuropsychologic evaluation in all patients. Oral examination performed by a dentist. Detailed skin examination in FA and TBD. Other evaluations as needed (such as gastrointeistinal endoscopy or nasolaryngoscopy screening) | ||
| • Hematologic evaluation: disease phase (single or multilineage cytopenias, MDS, AML). CBC, fetal hemoglobin, eADA (Diamond-Blackfan anemia), α-fetoprotein, bone marrow aspirate, biopsy, cytogenetics and flow cytometry. FISH for chromosomes 3 and 7 in FA. | ||
| • Previous transfusions (iron overload): Check number of transfusions, chelation history and ferritin levels. Consider T2* MRI to determine liver/heart iron overload. Check for alloimmunization and presence of DSA. | ||
| • Prior use of androgens: describe type, duration and dose. Check for signs of virilization, growth problems, and liver dysfunction. Check abdominal ultrasound, liver function, lipid metabolism, and bone age. | ||
| • Prior use of steroids: describe type, duration, and dose. Check for signs of Cushing’s syndrome; hyperglycemia, hypertension, metabolic syndrome, avascular necrosis, and adrenal insufficiency. | ||
| • For adults: special attention to genitourinary and gynecologic issues. Address fertility and options available for cryopreservation before HCT. Aggressive cancer screening is very important in this population. | ||
| • Lifestyle evaluation: Recommend complete abstinence from smoking and alcohol especially for FA and TBD, good oral hygiene, sunscreen use, healthy diet, and exercise. | ||
| Disease-specific pretransplant evaluation | ||
| FA | DC | DBA |
| Address congenital abnormalities (head, heart, skeletal, genitourinary, and gastrointestinal). | Address congenital abnormalities. Brain MRI is indicated in HH and Revesz and/or development delay | Address congenital abnormalities |
| Brain MRI is indicated for children with multiple birth defects (pituitary gland evaluation) | Immune abnormalities are common in early-onset cases. | Prolonged use of steroids may cause pathologic fractures, avascular necrosis, cataracts, growth retardation, hypertension, and diabetes |
| Avascular necrosis of the hips | ||
| Attention to endocrine problems such as short stature, thyroid function, glucose, gonadal function, and lipid metabolism | Attention to PFTs and SpO2 before HCT. Chest CT scan when indicated. | Iron overload is the major complication leading to heart and liver hemosiderosis, insulin-dependent diabetes mellitus, hypothyroidism, and delayed puberty |
| Postpuberty: Fertility evaluation | Check for signs of pulmonary and liver fibrosis and arteriovenous malformations in the lung, liver, and gastrointestinal tract, especially in adult patients | |
| • Complete family and patient’s history with careful evaluation of physical findings according to disease type. | ||
| • Multidisciplinary team: Visual, hearing, endocrine, nutritional, and neuropsychologic evaluation in all patients. Oral examination performed by a dentist. Detailed skin examination in FA and TBD. Other evaluations as needed (such as gastrointeistinal endoscopy or nasolaryngoscopy screening) | ||
| • Hematologic evaluation: disease phase (single or multilineage cytopenias, MDS, AML). CBC, fetal hemoglobin, eADA (Diamond-Blackfan anemia), α-fetoprotein, bone marrow aspirate, biopsy, cytogenetics and flow cytometry. FISH for chromosomes 3 and 7 in FA. | ||
| • Previous transfusions (iron overload): Check number of transfusions, chelation history and ferritin levels. Consider T2* MRI to determine liver/heart iron overload. Check for alloimmunization and presence of DSA. | ||
| • Prior use of androgens: describe type, duration and dose. Check for signs of virilization, growth problems, and liver dysfunction. Check abdominal ultrasound, liver function, lipid metabolism, and bone age. | ||
| • Prior use of steroids: describe type, duration, and dose. Check for signs of Cushing’s syndrome; hyperglycemia, hypertension, metabolic syndrome, avascular necrosis, and adrenal insufficiency. | ||
| • For adults: special attention to genitourinary and gynecologic issues. Address fertility and options available for cryopreservation before HCT. Aggressive cancer screening is very important in this population. | ||
| • Lifestyle evaluation: Recommend complete abstinence from smoking and alcohol especially for FA and TBD, good oral hygiene, sunscreen use, healthy diet, and exercise. | ||
| Disease-specific pretransplant evaluation | ||
| FA | DC | DBA |
| Address congenital abnormalities (head, heart, skeletal, genitourinary, and gastrointestinal). | Address congenital abnormalities. Brain MRI is indicated in HH and Revesz and/or development delay | Address congenital abnormalities |
| Brain MRI is indicated for children with multiple birth defects (pituitary gland evaluation) | Immune abnormalities are common in early-onset cases. | Prolonged use of steroids may cause pathologic fractures, avascular necrosis, cataracts, growth retardation, hypertension, and diabetes |
| Avascular necrosis of the hips | ||
| Attention to endocrine problems such as short stature, thyroid function, glucose, gonadal function, and lipid metabolism | Attention to PFTs and SpO2 before HCT. Chest CT scan when indicated. | Iron overload is the major complication leading to heart and liver hemosiderosis, insulin-dependent diabetes mellitus, hypothyroidism, and delayed puberty |
| Postpuberty: Fertility evaluation | Check for signs of pulmonary and liver fibrosis and arteriovenous malformations in the lung, liver, and gastrointestinal tract, especially in adult patients | |
These are disease-specific evaluations. All patients with IBMFS should undergo other regular pretransplant evaluation according to each BMT center. A more complete description of system involved in patients with IBMFS can be found in Alter.1 AML, acute myeloid leukemia; BMT, bone marrow transplantation; CBC, complete blood counts with differential and reticulocytes; CT, computed tomography; DSA, donor-specific antibodies; eADA, erythrocyte adenosine deaminase; FISH, fluorescent in situ hybridization; HH, Hoyeraal-Hreidarsson syndrome; MDS, myelodysplastic syndrome; MRI, magnetic resonance imaging; SpO2, saturation of oxygen by pulse oximetry.
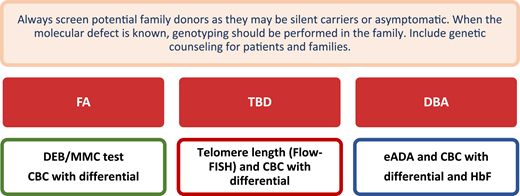
Screening of potential family donors. For a detailed discussion about diagnosis and genetic testing for IBMF and hematopoietic malignancy syndromes, the readers should refer to Furutani and Shimamura.2 DEB, diepoxybutane chromosome breakage test; Flow-FISH, flow cytometry with fluorescent in situ hybridization. CBC, complete blood counts with reticulocytes; HbF, fetal hemoglobin.
Screening of potential family donors. For a detailed discussion about diagnosis and genetic testing for IBMF and hematopoietic malignancy syndromes, the readers should refer to Furutani and Shimamura.2 DEB, diepoxybutane chromosome breakage test; Flow-FISH, flow cytometry with fluorescent in situ hybridization. CBC, complete blood counts with reticulocytes; HbF, fetal hemoglobin.
Fanconi anemia
Clinical case 1
Patient 1 is a 14-year-old girl, and she presented with a history of mild anemia since the age of 5 treated with supportive care. Over the past months, her mother noticed progressive asthenia, petechiae, and oral bleeding. Physical examination was remarkable for short stature and 2 café au lait spots. Complete blood counts showed severe pancytopenia, and she received red blood cell and platelet transfusions. A bone marrow aspirate and biopsy confirmed severe aplastic anemia. She was worked-up for Fanconi anemia (FA) at a tertiary referral center, and the diagnosis was confirmed with a positive diepoxybutane chromosome breakage test.
FA, predominantly an autosomal recessive disease, is characterized by genomic instability, progressive bone marrow failure, increased frequency of birth defects, and a striking predisposition for myeloid leukemias and head and neck squamous cell carcinoma (SCC).3,6,9 HCT offers cure for the hematologic complications of FA and is indicated before onset of chronic transfusions, myelodysplasia (MDS), or acute myeloid leukemia (AML).6,8,10 Recent studies have demonstrated an overall survival (OS) of 80% to 90% for young patients transplanted for aplasia from matched related (MRD) or matched unrelated donors (MUD).6,11,12 For MRD transplantation, low-dose cyclophosphamide (CY) 20 to 40 mg/kg and fludarabine with or without antithymocyte globulin (ATG) is the most common regimen,6,8,12 and irradiation is not necessary.13 Our group has transplanted 91 FA patients in aplastic phase with MRD and CY 60 mg/kg with or without ATG; with a median follow-up of 7 years, 95% of patients were alive.14 Outcomes after HCT for patients with FA transplanted in aplastic phase from MRD have improved dramatically over the last 30 years, and major long-term complications are now mainly related to endocrinologic problems and the development of cancer.1,5,6,15
Should we give androgens before HCT while waiting for an alternative donor?
We recommend androgens at the lowest possible dose (danazol, 2-5 mg/kg) while waiting for an unrelated donor. For patients in aplastic phase, androgen therapy (oxymetholone/danazol) leads to hematologic responses in up to 80% of patients.16-18 Patients who respond to androgens may remain stable for years, enabling advances in donor selection and transplantation procedures.6,19 In our daily practice, the decision to transplant a patient who has achieved a good hematologic response is never easy and usually depends on whether a well-matched alternative donor is available. Some patients or families may opt for continuing the use of androgens and delaying the transplant when only mismatched donors are available because of the higher risk related to these transplants. However, it is important to emphasize that androgens do not prevent the development of clonal evolution, the response may be transitory, patients may be older, and the donor may not be available when transplantation is needed. Side effects are also considerable, including dyslipidemia, abnormal liver function, liver adenomas, accelerated growth, and virilization, especially in young girls.16-18 Regular assessment with ultrasonography, liver function, and lipid metabolism are necessary before transplant.3,4,16-18
What are the results and specific problems related to HCT in FA patients?
Patient 1 did not have a suitably matched related donor, and while awaiting an unrelated donor search, she received androgens and achieved a partial response. A matched unrelated donor was not found, and she developed signs of virilization and elevated liver enzymes. She was then considered for a mismatched unrelated donor transplantation or a haploidentical transplant.
The use of alternative donors has been historically associated with a lower survival because of high incidence of rejection and regimen-related toxicity, as well as GVHD.6,8,10,11 Pivotal studies were able to identify changes that would enhance engraftment, decrease transplant-related mortality, and improve survival.10 These factors included the incorporation of fludarabine in preparatory regimens, the use of in vivo or in vitro T-cell depletion, and better HLA typing and supportive care.8,11,20 Young patients transplanted in aplastic phase from MUD receiving nonirradiation regimens have a survival that is now comparable to matched sibling donor (MSD) transplantation.6,7,11
For patients without a MRD or MUD, in vitro or in vivo T-cell depletion of grafts from a mismatched donor is an attractive option. In vitro T-cell depletion of the graft is effective in eliminating severe acute and chronic GVHD; however, this requires techniques that are not widely available.10,11 Using haploidentical donors with the posttransplant cyclophosphamide platform is associated with a 2-year OS of 80% to 90%, but GVHD is higher, and longer follow-up is still needed to better identify late outcomes.20,21 The use of cord blood transplantation has been associated with decreased rates of neutrophil engraftment and higher mortality, but using a fludarabine-based RIC regimen and selecting a unit with high cell dose and no more than one mismatch may help to overcome these complications.10,22 In places where it is available and when feasible, families with known gene mutations have an option to go for in vitro fertilization with preimplantation genetic diagnosis to select unaffected HLA matched embryos and subsequent cord blood transplantation for their child with FA. However, this is very expensive and can be physically and emotionally demanding for many families.22,23
Clinical case 1 continued
Because of the lack of complete response and unacceptable side effects related to androgens, patient 1 received a haploidentical HCT from her unaffected brother with fludarabine, ATG, and total body irradiation 200 cGy conditioning, followed by GVHD prophylaxis with posttransplant cyclophosphamide (total dose, 50 mg/kg), cyclosporine, and intravenous mycophenolate mofetil.8 Posttransplant course was complicated by grade III mucositis, cytomegalovirus reactivation, hemorrhagic cystitis, and mild oral chronic GVHD. At the age of 21, she became pregnant and delivered a healthy baby.
Age is an important predictor for survival after HCT. In a study of 199 adults with FA, 46% had clonal disease, and the 3-year OS for the whole group was 36%.24 Mortality secondary to transplant-associated complications was high (51%) and primarily caused by infections and GVHD. In the subset transplanted after 2000 with MRD, 3-year survival was 84%. Second malignancy remains an important complication, and transition from pediatric to adult care is challenging with many patients lost to follow-up in adulthood.24
Treatment of patients with MDS and AML can be very challenging.6,10,24,25 As expected, those transplanted in remission have a better survival, but the risk of giving pretransplant chemotherapy is high and may lead to excessive toxicity and prolonged aplasia. A sequential approach using a FLAG regimen followed by RIC seems promising, but the numbers are still small to draw any conclusions.25 Recently, the European Society for Blood and Marrow Transplantation group reviewed the data on 74 patients, most transplanted with MDS (n = 35) or had leukemia (n = 35). The 5-year OS was 42% with a relapse rate of 40% at 5 years.26
Long-term follow-up is essential to detect complications related to the disease or treatment. Endocrine abnormalities are the most frequent problems and may include growth hormone deficiency, thyroid dysfunction, dyslipidemia, hypogonadism, glucose intolerance, insulin resistance, or diabetes.5,15,27 All males are infertile, and although fertility is low in females, successful pregnancies have been reported as described in our clinical case.5,15 FA patients continue to be at risk for developing cancer after transplant, especially head and neck SCC and the use irradiation-containing regimens and/or GVHD may increase the incidence of this complication. Lifelong aggressive surveillance for early detection of cancer is recommended because the treatment options in FA are limited, and complications related to chemotherapy and radiotherapy are frequent.1,5,9,15,28,29
Telomere biology disorders
Clinical case 2
Patient 2 is an 11-year-old boy presenting with development delay and progressive bone marrow failure who was eventually diagnosed with dyskeratosis congenita (TINF2 gene mutation). He received a MUD transplant at age 13 with cyclophosphamide, fludarabine, ATG conditioning, and GVHD prophylaxis with cyclosporine and methotrexate. Three years later, he presented with progressive dyspnea. Physical examination was remarkable for multiple dental caries, cyanosis, digital clubbing, mild splenomegaly, and oxygen saturation of 87% in room air. High-resolution chest computed tomography scan showed no signs of parenchymal lung disease. However, intrapulmonary shunting was detected by contrast-enhanced echocardiography with agitated saline, suggesting hepatopulmonary syndrome (HPS). The patient was started on androgens (danazol) and supplemental oxygen without clinical response. He subsequently presented with seizures, fever, and a brain abscess attributed to a bacterial embolus consequent to right-to-left shunting and died despite appropriate management.
During the past decades, the discovery of germline mutations in 14 genes involved in the telomere maintenance has led to the definition of a group of disorders called telomere biology disorders (TBDs).4 There is excessive telomere attrition, with very short telomeres, less than the first percentile for age measured in leukocytes subsets. Dyskeratosis congenita (DC) represents the prototype of TBD and is characterized by bone marrow failure and the classical triad of reticular skin pigmentation, oral leukoplakia, and nail dystrophy. Most patients develop cytopenia at least in 1 lineage by the age of 40.4 The most severe variants present in childhood and include Hoyeraal-Hreidarsson syndrome (cerebellar hypoplasia, microcephaly, developmental delay, immunodeficiency) and Revesz syndrome (bilateral exudative retinopathy).4 The TBD patient may also present with liver cirrhosis and pulmonary fibrosis and is at a lifetime higher risk for MDS, AML, and solid tumors.3,4,9 Recent studies have focused on a group of vascular abnormalities including pulmonary arteriovenous malformations and gastrointestinal telangiectasias that are a cause of substantial morbidity and mortality.1,4,30
Treatment with androgens may improve the marrow function in most patients, and the use of danazol is preferred because it has fewer virilizing symptoms. Although side effects are similar to the ones described for FA, patients with DC may be more susceptible to liver complications, and the risk of splenic peliosis and splenic rupture is increased when growth factors are used simultaneously.4,17,31
What are the results and specific problems related to HCT in TBD patients?
HCT remains the only curative treatment of the hematologic complications, but survival is limited by a high incidence of both early and long-term complications.1,4,5,7,32 Understanding the wide spectrum of clinical manifestations (pulmonary, gastrointestinal, liver, hematologic, neurologic, ophthalmic, and immunologic abnormalities) is important to systematically evaluate each patient before and after transplant (Tables 1 and 2).1,4,15,30
Posttransplant evaluation of patients with IBMFS
| • General recommendations: Maintain healthy diet, regular exercise, good oral hygiene, and sunscreen use. | ||
| • Recommend complete abstinence from alcohol and smoking (including vaping). | ||
| • Ensure HPV vaccination for all patients. | ||
| • Posttransplant evaluation is a multidisciplinary teamwork. Consider organizing a LTFU clinic in collaboration with other specialists (endocrinologists, dentists, head and neck oncologists, gynecologists, and others). | ||
| • Consider working together with family associations and organizing regional/national family meetings as this may help patients, increase disease awareness, and improve research | ||
| • Attention to neurocognitive issues, especially in patients with development delays | ||
| • Psychologic evaluation and psychologic support | ||
| • Address visual and hearing problems as they may impact the learning process and decrease academic achievements and quality of life | ||
| • Annual liver, kidney, and gastrointestinal evaluation. Cardiac and pulmonary evaluation every other year, except for DC patients (see below) | ||
| • Annual endocrine evaluation: growth assessment, glucose, lipid metabolism. Assess gonadal function and bone mineral density | ||
| • For postpubertal female patients: annual gynecologic evaluation. Discuss fertility options. | ||
| • For male patients: gonadal function and spermogram | ||
| • Iron overload: Check ferritin levels within 6 mo to 1 y of transplantation. Consider T2* MRI to determine liver iron overload. Phlebotomy is the first choice of treatment; second is desferasirox | ||
| • GVHD increases the risk of cancer after HCT for all patients with IBMFS. Treatment of GVHD with steroids may be associated with metabolic syndrome, diabetes, avascular necrosis, and adrenal insufficiency. | ||
| • Aggressive cancer surveillance. Cancer risk increases as patients get older and in the presence of GVHD | ||
| • Dermatologic evaluation: skin cancer screening every 6-12 mo | ||
| • Oral examination performed by a dentist every 6-12 mo. Encourage monthly oral self-examination | ||
| Disease-specific posttransplant complications | ||
| FA | DC | DBA |
| Endocrinologic problems are very frequent after HCT, including thyroid dysfunction, fertility, hypogonadism, and growth hormone (GH) deficiency. | Pulmonary and liver complications are the major problems after HCT | Iron overload is the major complication after HCT, and LTFU problems include diabetes, delayed puberty, and hypothyroidism. |
| Short stature can be treated with GH after 6 mo of HCT, if GH deficiency is confirmed.27 | Perform annual PFTs and check SpO2 and signs of pulmonary and/or liver fibrosis and arteriovenous malformations (lung, liver, and gastrointestinal tract) after HCT 30,35 | Other problems include those related to chronic steroid use and fertility issues.38,39 |
| Cancer risk9 | ||
| Skin SCC and basal cell carcinoma | Skin SCC and basal cell carcinoma | Colorectal carcinoma |
| Head and neck and anorectal and vulvar SCC | Head and neck anorectal SCC | Osteogenic sarcoma |
| Esophagus, breast, and brain cancer | Esophagus, stomach, and lung cancer | |
| • General recommendations: Maintain healthy diet, regular exercise, good oral hygiene, and sunscreen use. | ||
| • Recommend complete abstinence from alcohol and smoking (including vaping). | ||
| • Ensure HPV vaccination for all patients. | ||
| • Posttransplant evaluation is a multidisciplinary teamwork. Consider organizing a LTFU clinic in collaboration with other specialists (endocrinologists, dentists, head and neck oncologists, gynecologists, and others). | ||
| • Consider working together with family associations and organizing regional/national family meetings as this may help patients, increase disease awareness, and improve research | ||
| • Attention to neurocognitive issues, especially in patients with development delays | ||
| • Psychologic evaluation and psychologic support | ||
| • Address visual and hearing problems as they may impact the learning process and decrease academic achievements and quality of life | ||
| • Annual liver, kidney, and gastrointestinal evaluation. Cardiac and pulmonary evaluation every other year, except for DC patients (see below) | ||
| • Annual endocrine evaluation: growth assessment, glucose, lipid metabolism. Assess gonadal function and bone mineral density | ||
| • For postpubertal female patients: annual gynecologic evaluation. Discuss fertility options. | ||
| • For male patients: gonadal function and spermogram | ||
| • Iron overload: Check ferritin levels within 6 mo to 1 y of transplantation. Consider T2* MRI to determine liver iron overload. Phlebotomy is the first choice of treatment; second is desferasirox | ||
| • GVHD increases the risk of cancer after HCT for all patients with IBMFS. Treatment of GVHD with steroids may be associated with metabolic syndrome, diabetes, avascular necrosis, and adrenal insufficiency. | ||
| • Aggressive cancer surveillance. Cancer risk increases as patients get older and in the presence of GVHD | ||
| • Dermatologic evaluation: skin cancer screening every 6-12 mo | ||
| • Oral examination performed by a dentist every 6-12 mo. Encourage monthly oral self-examination | ||
| Disease-specific posttransplant complications | ||
| FA | DC | DBA |
| Endocrinologic problems are very frequent after HCT, including thyroid dysfunction, fertility, hypogonadism, and growth hormone (GH) deficiency. | Pulmonary and liver complications are the major problems after HCT | Iron overload is the major complication after HCT, and LTFU problems include diabetes, delayed puberty, and hypothyroidism. |
| Short stature can be treated with GH after 6 mo of HCT, if GH deficiency is confirmed.27 | Perform annual PFTs and check SpO2 and signs of pulmonary and/or liver fibrosis and arteriovenous malformations (lung, liver, and gastrointestinal tract) after HCT 30,35 | Other problems include those related to chronic steroid use and fertility issues.38,39 |
| Cancer risk9 | ||
| Skin SCC and basal cell carcinoma | Skin SCC and basal cell carcinoma | Colorectal carcinoma |
| Head and neck and anorectal and vulvar SCC | Head and neck anorectal SCC | Osteogenic sarcoma |
| Esophagus, breast, and brain cancer | Esophagus, stomach, and lung cancer | |
These are disease-specific recommendations. All patients with IBMFS should undergo other regular posttransplant evaluation according to published guidelines.1,15 LTFU, long-term follow-up; SpO2, oxygen saturation by pulse oximetry.
In a recent study analyzing the outcome of 94 patients, most were transplanted after 2000 (84%) and from MUD (49%). All patients received a RIC regimen, and the 5- and 10-year OS was 59% and 30%, respectively.32 Consistent with another report, most early deaths and graft failure occurred after mismatched donor transplantations.33 Late mortality was attributed mainly to liver and pulmonary fibrosis and other vascular complications, most likely related to the underlying disease.32,33 To address the benefit of less toxic preparatory regimens, a prospective multi-institutional HCT clinical trial for patients with TBD is currently testing whether a radiation and alkylator-free regimen can lead to successful and sustained engraftment and fewer long-term complications (clinicaltrials.gov #NCT01659606). Our HCT center has transplanted 28 patients with TBD in aplastic phase over a 20-year period.34 Most received a radiation-free RIC regimen and bone marrow from MUD (46%). With a median follow-up of 6 years, the 5-year OS was 53%. However, only 2 of 8 patients transplanted from mismatched donors are alive. We observed an unusually high frequency of vascular complications including severe hepatic VOD (n = 1), recurrent gastrointestinal bleeding (n = 1; Revesz syndrome), and HPS (n = 5). Only 1 patient with HPS is alive, and none had abnormal pulmonary function tests (PFTs) before transplant.34 Patients with HPS may also develop liver dysfunction, but the diagnosis usually requires a liver biopsy to detect intrahepatic arteriovenous malformations and regenerative nodular hyperplasia.1,4,30 In a recent report, patients with abnormal baseline PFT had a higher risk for significant pulmonary disease. In that study, the median time to pulmonary complications after transplant was 4.7 years,35 very similar to what we observed in our cohort (5 years).34 PFT and an evaluation to detect vascular abnormalities should be performed regularly before and after HCT, and with HPS, a complete hepatic evaluation.1,4,35 Currently there is no effective treatment of pulmonary fibrosis, HPS, and liver cirrhosis other than solid organ transplantation.4,35 Because these complications impact survival after HCT, a multidisciplinary team approach is essential for early detection of liver and lung problems. The risk of cancer is also high, and the most common types are head and neck and anogenital SCC; life-long screening is recommended.4,9,15
Can we do anything to prevent disease progression after transplant?
One prospective trial in patients with TBD demonstrated telomere elongation and no significant decrease in lung function during danazol administration, suggesting that the use of danazol could slow down the progression of pulmonary fibrosis.31 Although these results are provocative, there is no confirmed role for the use of androgens in preventing disease progression in nonhematopoietic tissues.4 Other approaches to prevent disease complications in the future may include the use of small molecules that specifically restore telomere maintenance in patient-derived stem cells, exploring the use of WNT pathway agonists such as lithium, and the use of small peptides derived from dyskerin that have a role in decreasing oxidative stress, DNA damage, and cell senescence.4,36,37
Diamond-Blackfan anemia
Clinical case 3
Patient 3 is a 4-year-old boy with a confirmed diagnosis of Diamond-Blackfan anemia (DBA) and was referred for an MSD transplantation. The donor was his healthy unaffected 8-year-old brother with normal blood counts. Patient 3 was treated irregularly with high-dose steroids since he was 8 months old and needed monthly red blood cell transfusions with inadequate chelation. Physical examination was remarkable for signs of Cushing syndrome, growth failure, and thumb abnormalities. He received a myeloablative conditioning (MAC) regimen with busulfan, fludarabine, ATG, and GVHD prophylaxis with cyclosporine and methotrexate. The transplant was uneventful, and the patient is alive 5 years after transplant. Major long-term sequelae include growth failure and iron overload.
DBA represents a group of disorders with a defective ribosome biogenesis and is characterized by anemia with reticulocytopenia, congenital abnormalities, and an increased predisposition to cancer. In the classical form, patients present in the first year of life with a macrocytic anemia and a normocellular marrow with selective paucity of red cell precursors. Congenital abnormalities are frequent, and nearly half of the patients have craniofacial, skeletal, heart, and kidney abnormalities.38
The standard of care for DBA includes corticosteroids and chronic transfusions with adequate iron chelation. Although 60% to 80% of patients respond to an initial course of corticosteroids, less than 50% can be maintained with doses that are low enough to avoid long-term toxicity, and fewer than 20% achieve spontaneous remission.39 It is recommended to avoid steroids in the first year of life because of its deleterious effects on linear growth, and infants should be treated with regular blood transfusions. Chelation should be initiated when patients receive approximately 200-mL/kg red blood cell transfusions.39,40
HCT is the only curative option for the hematologic manifestations of DBA and is recommended for patients who fail to respond to corticosteroids and/or need chronic transfusion. We and others consider treatment failure as needing corticosteroids (dose >0.3 mg/kg per day) to maintain hemoglobin >8 g/dL.7,39 Although not frequent, HCT may also be indicated for hematologic malignancy or aplastic anemia.7,40 It is essential to screen potential family donors because they may be silent carriers or asymptomatic.38,39 In our clinical case 3, the patient’s genotype was known, and it was easy to screen the donor. However, when this is not available, donors should have a complete blood count, reticulocyte count, and fetal hemoglobin. Although erythrocyte adenosine deaminase activity is elevated in most patients, the test is available only in a few countries around the world.38
What are the results and specific problems related to HCT in DBA patients?
Many patients present with complications related to the use of corticosteroids, Cushing syndrome (diabetes, hypertension, central adiposity), and chronic transfusion: iron overload (liver and heart), alloimmunization, and anti-HLA antibodies. Recent studies report excellent OS (90%-100%) after MRD HCT for patients <10 years of age.40,41 Current guidelines recommend a busulfan or treosulfan with fludarabine and serotherapy (MAC regimen). Engraftment rates are high, and severe acute and chronic GvHD rates are low.7 A MUD HCT is also associated with similar results.40 However, mismatched unrelated donor HCT leads to high graft failure and GVHD. In a recent report from Brazil (n = 44), the 5-year OS after MSD HCT was 80%, for MUD was 73%, and for mismatched unrelated donors was 29%.42 Common causes of death included graft failure and infections. As with other IBMFS, iron overload from chronic transfusions adds to the burden of morbidity post-HCT and requires intermittent phlebotomy. Aggressive iron chelation before and after transplant is necessary to reduce cardiac iron overload because deaths from cardiac causes have occurred as late as 5 to 7 years after HCT.1,15 Patients without venous access may tolerate oral iron chelators such as desferasirox.1,15,17,39 The use of a MAC regimen may lead to infertility, and fertility preservation (when available) should be discussed upfront. DBA is a cancer predisposition syndrome, and the most common neoplasias include colon and genitourinary cancers, osteogenic sarcoma, and myeloid malignancies. Guidelines for long-term follow-up in patients with IBMFS have been recently updated.1,15,39
Other IBMFS with hematopoietic malignancy predisposition syndromes.
Severe congenital neutropenia is characterized by recurrent life-threatening bacterial infections, chronic neutropenia from birth, and predisposition to hematologic malignancies. Treatment with granulocyte colony-stimulating factor is the standard of care, and HCT is recommended for patients who are not responsive to high doses of daily granulocyte colony-stimulating factor or in the presence of clonal evolution. Young patients transplanted from matched related or unrelated donors with myeloablative preparatory regimens have the best outcomes.7,43 Shwachman-Diamond syndrome is a recessive disorder characterized by bone marrow failure, exocrine pancreatic insufficiency, and skeletal abnormalities. Malignant transformation is frequent, ranging between 5% and 25%. The indications for transplant are worsening cytopenias and transformation into MDS and AML.7 Recent studies from Europe and the United States showed a 5-year overall survival of 70% for patients transplanted in marrow failure and a dismal outcome for those transplanted with MDS or AML.44,45
Recent advances in genomic evaluation with next-generation sequencing can identify many suspected IBMFS with atypical presentations that remain undiagnosed after an initial workup.2,3 Improving genetic diagnosis comes with a price and may delay referral to transplantation because these techniques are not routinely available worldwide. The balance between the urgency to proceed to transplantation and the need to confirm the diagnosis in a patient with suspected IBMFS to ensure that a healthy related donor is selected is still a matter of debate, especially in developing countries. Until we establish international partnerships, a great number of patients will remain underdiagnosed, and prognosis after transplantation will not be adequately established. Diseases such as congenital amegakaryocytic thrombocytopenia, GATA2 deficiency, MECOM syndrome, CTLA4 haploinsufficiency, and SAMD9 are being increasingly diagnosed because of precision genomics, and transplant outcomes for these rare indications are shown in Table 3.46-50
HCT for other hematopoietic malignancy predisposition syndromes
| Disease and reference | No. pts, median age | Disease phase | Prep regimen | Donor type stem cell source | GVHD | TRM | OS | Follow-up | Comments |
|---|---|---|---|---|---|---|---|---|---|
| CAMT46 | 63 pts | NR | MAC 82% | MRD or MUD > 80% | A-GVHD: II-IV 13%. | 13% at 3 y | 5-y OS: 76% | 1,7 y (0.1-14) | No difference in OS according to donor or stem cell source |
| 7 y (0.5-17) | BM (n = 31); PB (n = 21); UCB (n = 11) | Extensive C-GVHD: 6.3% | |||||||
| GATA247 | 15 pts | MDS (n = 12) | 73% CY + TBI | MRD or MUD 53% | A-GVHD: II- IV: 33% | 20% at 1 y | 5-y OS: 65% 5-y DFs:51 | 5,0 y (1-8.5) | Neurological toxicities and thrombotic events more frequent after GATA2 transplants |
| 15 y (5-20) | AL (n = 3) | BM (n = 11) and UCB (n = 4) | Extensive C-GVHD 26% | ||||||
| MECOM49 | 10 pts | Progressive BMF | NR | MRD or MUD: 9 pts | NR | 3 pts died | 7 pts alive | NR | Sensorineural deafness, kidney, heart, and skeletal defects |
| 0.7 y (0.2-13) | BM (n = 9) and UCB (n = 1) | ||||||||
| MECOM48 | 6 pts | Progressive BMF | NR | NR | NR | 2 deaths from cardiac problems | 4 pts alive | 10 mo | 4 pts are alive with no major complications after HCT |
| 1.3 y (0.5-3) | |||||||||
| ERCC6L248 | 3 pts | BMF (n = 2) | NR | NR | NR | 1 death EBV lymphoma | 2 pts alive | 1 and 14 y | 2 pts are alive with no major complications after HCT |
| 13; 14; 22 y | MDS (n = 1) | ||||||||
| SAMD9: 4 pts | BMF (n = 3) | NR | NR | NR | 2 deaths | 8 pts alive | 4 y (0.1-7) | SAMD9: 3 pts alive and well | |
| SAMD9 | SAMD9L: 6 pts | ||||||||
| SAMD9L48 | 3 y (1.5-33) | MDS (n = 7) | SAMD9L: 5 pts alive, one with neurological complication | ||||||
|
|
|
|
|
|
| 10 pts alive | 3.1 y (0.1-14) |
|
| Disease and reference | No. pts, median age | Disease phase | Prep regimen | Donor type stem cell source | GVHD | TRM | OS | Follow-up | Comments |
|---|---|---|---|---|---|---|---|---|---|
| CAMT46 | 63 pts | NR | MAC 82% | MRD or MUD > 80% | A-GVHD: II-IV 13%. | 13% at 3 y | 5-y OS: 76% | 1,7 y (0.1-14) | No difference in OS according to donor or stem cell source |
| 7 y (0.5-17) | BM (n = 31); PB (n = 21); UCB (n = 11) | Extensive C-GVHD: 6.3% | |||||||
| GATA247 | 15 pts | MDS (n = 12) | 73% CY + TBI | MRD or MUD 53% | A-GVHD: II- IV: 33% | 20% at 1 y | 5-y OS: 65% 5-y DFs:51 | 5,0 y (1-8.5) | Neurological toxicities and thrombotic events more frequent after GATA2 transplants |
| 15 y (5-20) | AL (n = 3) | BM (n = 11) and UCB (n = 4) | Extensive C-GVHD 26% | ||||||
| MECOM49 | 10 pts | Progressive BMF | NR | MRD or MUD: 9 pts | NR | 3 pts died | 7 pts alive | NR | Sensorineural deafness, kidney, heart, and skeletal defects |
| 0.7 y (0.2-13) | BM (n = 9) and UCB (n = 1) | ||||||||
| MECOM48 | 6 pts | Progressive BMF | NR | NR | NR | 2 deaths from cardiac problems | 4 pts alive | 10 mo | 4 pts are alive with no major complications after HCT |
| 1.3 y (0.5-3) | |||||||||
| ERCC6L248 | 3 pts | BMF (n = 2) | NR | NR | NR | 1 death EBV lymphoma | 2 pts alive | 1 and 14 y | 2 pts are alive with no major complications after HCT |
| 13; 14; 22 y | MDS (n = 1) | ||||||||
| SAMD9: 4 pts | BMF (n = 3) | NR | NR | NR | 2 deaths | 8 pts alive | 4 y (0.1-7) | SAMD9: 3 pts alive and well | |
| SAMD9 | SAMD9L: 6 pts | ||||||||
| SAMD9L48 | 3 y (1.5-33) | MDS (n = 7) | SAMD9L: 5 pts alive, one with neurological complication | ||||||
|
|
|
|
|
|
| 10 pts alive | 3.1 y (0.1-14) |
|
A-GVHD, acute GVHD; AL, acute leukemias; BM, bone marrow; BMF, bone marrow failures; CAMT, congenital amegakaryocytic thrombocytopenia; C-GVHD, chronic GVHD; CY, cyclophosphamide; DAH, diffuse alveolar hemorrhage; NR, not reported; pts, patients; TBI, total body irradiation; UCB, unrelated cord blood.
Conclusions
Modifying well-known risk factors has resulted in substantial reduction in early mortality after HCT, thus extending survival into adulthood. An adequate selection and evaluation of patients before transplant, use of bone marrow from well-matched donors, and avoiding irradiation-containing preparatory regimens are universal recommendations for these patients. Screening of family members is essential to exclude affected individuals as potential donors. For all IBMFS HCT will not correct nonhematologic manifestations related to these diseases. The decision to proceed to transplant is complex, and all aspects of the transplant process should be considered, including but not limited to the phase of the disease, safety and quality of blood transfusions, pretransplant comorbidities, availability of techniques to manipulate the graft, and a capacity for adequate posttransplant care that includes long-term follow-up. IBMFS are rare, and international collaborative efforts are needed to advance the care of these patients.
Acknowledgments
The author thanks mentor Ricardo Pasquini (Federal University of Parana, Curitiba, Brazil). Pasquini pioneered hematopoietic cell transplantation in Latin America, and through his mentorship, the author developed an interest in nonmalignant diseases. The authors also thanks Mary Eapen (Medical College of Wisconsin, Milwaukee, WI) for critical review of the manuscript.
Correspondence
Carmem Bonfim, Hospital de Clinicas–Federal University of Parana, 15 andar, Serviço de Transplante de Medula Óssea, Rua General Carneiro 181, Curitiba PR CEP 80-060-900, Brazil; e-mail: carmembonfim@gmail.com.
