

Abstract
Patients with Gaucher disease (GD), a rare autosomal recessive glycosphingolipid storage disease, commonly present to hematologists with unexplained splenomegaly, thrombocytopenia, anemia, and bone symptoms. Patients with GD may develop other manifestations, such as autoimmune thrombocytopenia, monoclonal gammopathy, multiple myeloma, or, even more rarely, other hematological malignancies; sometimes they are first diagnosed during an assessment of those disorders. Although the diagnosis and management of patients with GD have significantly evolved over the last 30 years, some patients remain poor responders to GD-specific therapy, needing novel and investigational therapies. Ideally, patients with GD, like patients with other rare diseases, should be managed by a multidisciplinary team expert with the diverse clinical manifestations and potential GD-related or -unrelated comorbidities. The hematology community should be knowledgeable regarding the presentation and the variety of hematologic complications and comorbidities associated with Gaucher disease.
Learning Objectives
Recognize the clinical features and methods of diagnosis of Gaucher disease (GD) while avoiding pitfalls
Review current and new therapies for GD
Learn that plasma cell dyscrasias are potential comorbidities in GD
Introduction
Gaucher disease (GD) is an autosomal recessive glycosphingolipid storage disease caused by mutations of the lysosomal enzyme glucocerebrosidase gene (GBA1), leading to the accumulation of the substrate glucocerebroside in the cells of the macrophage–monocyte system. It is 1 of the 2 most common lysosomal storage disorders and has an estimated frequency of 1:50 000 to 1:100 000 in the general population, but with a much higher prevalence (∼1:850) in the Ashkenazi Jewish population. More than 860 different mutations in GBA1 have been identified, explaining, in part, the great phenotypic heterogeneity that is a hallmark of GD. Early diagnosis is important for timely initiation of specific therapy before the development of irreversible, mainly skeletal complications and for prenatal diagnosis in subsequent pregnancies.1
Most presenting features are hematological, as are some of the associated diseases (comorbidities) that develop in patients with GD at a higher incidence than in the general population, including immune thrombocytopenia, plasma cell dyscrasias, and other hematological malignancies, making it all the more important for hematologists to be familiar with this disease even if uncommon. Table 1 lists the main comorbidities among patients with GD.
Comorbidities among patients with Gaucher disease
| GD related | ERT related | SRT related |
|---|---|---|
| Amyloidosis | Class effect: | Class effect: |
| Cholelithiasis | ASIA/reactive arthritis | GI symptoms (eg, diarrhea, heartburn) |
| Celiac disease | Diabetes mellitus type 2 | Headache |
| Hashimoto thyroiditis | Hyperlipidemia | Drug specific: |
| Hepatocellular carcinoma | Metabolic syndrome | Cardiotoxicity |
| Immune thrombocytopenia | NAFLD/NASH | Cognitive impairment |
| Liver cirrhosis | Polyneuropathy | Drug–drug interaction |
| Lymphomas | Pulmonary hypertension | NSVT |
| MGUS | Weight gain | Peripheral neuropathy |
| Multiple myeloma | Drug-specific: | Tremor |
| Myelodysplastic syndrome | Allergy/hypersensitivity | Weight loss |
| Parkinson disease | ||
| Portal hypertension | ||
| Pulmonary hypertension | ||
| Recurrent abortions | ||
| Uveitis | ||
| Vitamin B12 deficiency | ||
| To be determined: gout, hypothyroidism, vitamin D deficiency |
| GD related | ERT related | SRT related |
|---|---|---|
| Amyloidosis | Class effect: | Class effect: |
| Cholelithiasis | ASIA/reactive arthritis | GI symptoms (eg, diarrhea, heartburn) |
| Celiac disease | Diabetes mellitus type 2 | Headache |
| Hashimoto thyroiditis | Hyperlipidemia | Drug specific: |
| Hepatocellular carcinoma | Metabolic syndrome | Cardiotoxicity |
| Immune thrombocytopenia | NAFLD/NASH | Cognitive impairment |
| Liver cirrhosis | Polyneuropathy | Drug–drug interaction |
| Lymphomas | Pulmonary hypertension | NSVT |
| MGUS | Weight gain | Peripheral neuropathy |
| Multiple myeloma | Drug-specific: | Tremor |
| Myelodysplastic syndrome | Allergy/hypersensitivity | Weight loss |
| Parkinson disease | ||
| Portal hypertension | ||
| Pulmonary hypertension | ||
| Recurrent abortions | ||
| Uveitis | ||
| Vitamin B12 deficiency | ||
| To be determined: gout, hypothyroidism, vitamin D deficiency |
ASIA, autoimmune syndrome induced by adjuvant; bold, most common GD-related comorbidities; ERT, enzyme replacement therapy; GD, Gaucher disease; GI, gastrointestinal; MGUS, monoclonal gammopathy of undetermined significance; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NSVT, nonsustained ventricular tachycardia; SRT, substrate reduction therapy.
Case report part 1: the pre–enzyme replacement therapy era
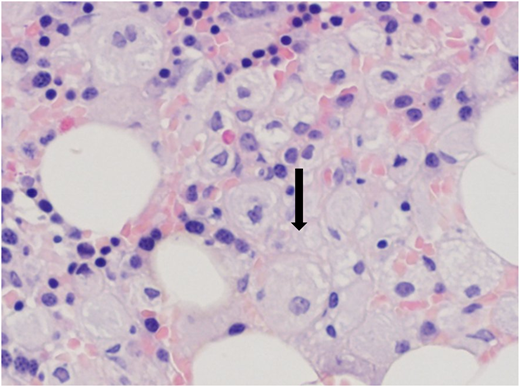
In 1974, a 19-year-old Ashkenazi Jewish woman was referred for hematological consultation for evaluation of menorrhagia and easy bruising. She had experienced epistaxis and blue markings since childhood, as well as a protuberant abdomen, but her parents were told that these were common in childhood. She had no history of bone pain or fractures. Her physical examination revealed a healthy-looking woman with multiple ecchymoses and marked splenomegaly. Bone marrow aspiration led to the diagnosis of GD based on the demonstration of engorged macrophages with cytoplasmic striations typical of “Gaucher cells” (Figure 1). With this benign diagnosis, no further workup or follow-up was recommended.
In the years before the introduction of enzyme replacement therapy (ERT) in 1991, the management of GD was purely symptomatic, including analgesics, splenectomy, and orthopedic surgery. Splenectomy was performed in severely symptomatic patients, frequently children with hypersplenism (the majority had thrombocytopenia with a bleeding diathesis, anemia, or uncommonly leukopenia), growth retardation, and even cachexia. However, because splenectomy removed the main storage organ, these surgeries often resulted in worsening of the hepatic and bony involvement, causing massive hepatomegaly (with or without liver function abnormalities) or cirrhosis, in addition to bone crises with pathological fractures and osteonecrosis. When this became recognized, splenectomies were deferred as long as possible for severely affected patients and avoided for milder patients.
The delayed diagnosis at age 19, about a decade after the onset of the first symptoms, is typical for GD, as it is for many rare disorders. In 2017, Mehta et al2 reported delays of 4 to 10 years in the United States and 0.6 to 26 years in the United Kingdom from first symptoms to diagnosis. In the case of our patient, who had a relatively mild clinical course, perhaps no harm was done. When she was diagnosed, in the premolecular era, carriers could not be accurately identified; genetic counseling was limited; and many Ashkenazi Jewish patients were advised to marry outside the Ashkenazi community.
Case report part 2: 24 years later, the era of GD-specific therapies
The patient presented at our Gaucher unit in 1998 at the age of 43 years and was married with 6 children. She was referred by the preoperative screening clinic (before repair of an umbilical hernia), where she was noted to have marked hepatosplenomegaly, pancytopenia, and abdominal ultrasonographic findings of multiple splenic lesions, suspicious for malignancy (Figure 2).
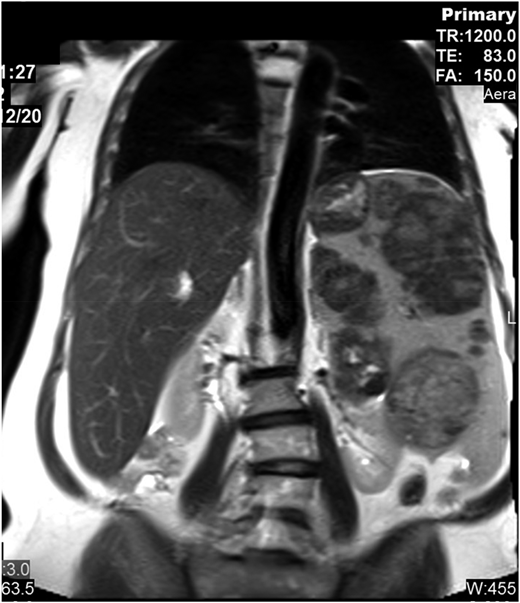
Abdominal magnetic resonance imaging showing hepatosplenomegaly with prominent intrasplenic lesions.
Abdominal magnetic resonance imaging showing hepatosplenomegaly with prominent intrasplenic lesions.
The diagnosis of GD was confirmed by reduced glucocerebrosidase activity and homozygosity of the N370S mutation in GBA1 (new nomenclature c.1226A>G; p.N409S). The patient’s family history was remarkable for 4 children with GD and a mother with Parkinson disease (PD), diagnosed at age 64.
Although recognized for almost 140 years, the natural history of untreated GD has not been fully appreciated. Many mildly affected and even asymptomatic patients receive lifelong ERT because their physicians erroneously assume that, without therapy, the disease will invariably deteriorate. However, GD progression (even in those with moderate to severe phenotypes) occurs during childhood, adolescence, and early adulthood with a tendency to stabilize later in life. In the majority of the mild or asymptomatic patients, GD remains unchanged for decades.3 Our moderately affected patient has remained stable for more than 2 decades. Intrasplenic (and less commonly intrahepatic) lesions—hypoechogenic, hyperechogenic, or mixed—are typical in GD, but for the inexperienced radiologist, they may mimic hematological malignancies, as in our patient.4
The gold standard for GD diagnosis is the demonstration of reduced β-glucocerebrosidase activity combined with whole GBA1 sequencing (to avoid pitfalls). Testing for specific biomarkers, such as chitotriosidase or glucosylsphingosine (LysoGb1), is helpful for diagnosis and for monitoring disease progression (in the untreated) and therapeutic response (for the treated patients).1 All 3 tests are conveniently done today as dry blood spots on filter papers. The key laboratory findings are delineated in Table 2.
Key laboratory findings
| Parameter | Description |
|---|---|
| Diagnosis | β-Glucosidase enzyme level, GBA1 mutations |
| Biomarkers | |
| Nonspecific | Elevated ferritin, acid phosphatase, and ACE |
| Reduced HDL cholesterol | |
| Specific | Elevated glucosylsphingosine (Lyso-Gb1), CCL18, and chitotriosidase |
| Hematology | |
| Cytopenia | Thrombocytopenia, anemia (less common), leukopenia (rare) |
| Coagulation | Low factor XI, coagulation abnormalities (less common), platelet dysfunction (adhesion, aggregation), abnormal thrombin generation |
| Gammopathies | Polyclonal, monoclonal |
| Free light chain abnormalities, serum immunofixation for detection of monoclonal immunoglobulins | |
| Biochemistry | Abnormal liver function tests |
| Reduced vitamin B12, folic acid, vitamin D |
| Parameter | Description |
|---|---|
| Diagnosis | β-Glucosidase enzyme level, GBA1 mutations |
| Biomarkers | |
| Nonspecific | Elevated ferritin, acid phosphatase, and ACE |
| Reduced HDL cholesterol | |
| Specific | Elevated glucosylsphingosine (Lyso-Gb1), CCL18, and chitotriosidase |
| Hematology | |
| Cytopenia | Thrombocytopenia, anemia (less common), leukopenia (rare) |
| Coagulation | Low factor XI, coagulation abnormalities (less common), platelet dysfunction (adhesion, aggregation), abnormal thrombin generation |
| Gammopathies | Polyclonal, monoclonal |
| Free light chain abnormalities, serum immunofixation for detection of monoclonal immunoglobulins | |
| Biochemistry | Abnormal liver function tests |
| Reduced vitamin B12, folic acid, vitamin D |
ACE, angiotensin-converting enzyme; CCL18, C-C motif chemokine ligand 18; HDL, high-density lipoprotein.
Although the N370S mutation, also known as the “common Jewish mutation,” is often associated with a mild or asymptomatic phenotype, various factors may negatively influence the clinical course of patients with this genotype, who may have more severe manifestations, including bony complications.5 With a 1:17 prevalence of this mutation in the Ashkenazi population and no premarital screening, our patient married an N370S carrier, and GD was diagnosed in 4 of her 6 children. They demonstrated the phenotypic diversity associated with this genotype: Her older son developed osteonecrosis of the hip and sacroiliac joints; one daughter had long-term thrombocytopenia before ERT; and the remaining 2 children are asymptomatic.
Another interesting aspect is her mother’s PD. PD is by far the most common GD-related comorbidity, and a family history of PD is described in ∼25% of patients. An increased risk for PD is also found among GD carriers with an increased risk of 3- to 15-fold relative to the general population, according to the severity of the mutation.6 On the basis of the assumptive underlying pathophysiology (ie, “loss of function” [haploid insufficiency] related to excess of the substrate glucocerebroside in the dopaminergic neurons versus “gain of function,” the impact of the misfolded mutant glucocerebrosidase on the aggregation of α-synuclein), there are several clinical trials in GBA1-related PD using substrate reduction therapy (SRT) (venglustat), in vivo adeno-associated virus 9–based gene therapy, or pharmacological chaperones (PCs).7,8 A family history of PD in a patient presenting with splenomegaly and/or thrombocytopenia should suggest GD in the differential diagnosis.
Case report part 3: poor response to specific therapies
With marked splenomegaly, severe thrombocytopenia (<50 × 109/L), and anemia, the patient fulfilled the criteria for ERT but was reluctant to receive IV imiglucerase (the only ERT available in 1998) and elected to join the clinical trial of a then-novel oral SRT (miglustat). After 12 months on miglustat, which she tolerated, she had no significant improvements in any of the key disease features, and she chose to remain untreated for the next 7 years.
In 2007, she joined the pivotal trial of taliglucerase alfa, the first plant cell–derived human recombinant ERT. Again, she experienced no adverse effects but no efficacy either. She remained off therapy for 4 more years before starting velaglucerase alfa, which she received for 6 years, still with no improvement despite increased dosages from 15 to 60 U/kg every other week.
The first ERT has revolutionized the management of patients with GD and opened the door for the development of many other treatment modalities for rare diseases in general. It was harvested from a huge volume of human placentas and in 1994 was replaced by the human recombinant imiglucerase, which was until 2010 the only ERT available worldwide. Due to its prohibitive cost, many countries other than the United States have defined criteria for treatment reimbursement, typically covering the more severely affected adult patients as well as symptomatic children. In Israel, patients who fulfill the government criteria usually receive a lower dosage than patients in the United States.9 Our patient joined the seminal clinical trial of the oral SRT miglustat (the iminosugar N-butyldeoxynojirimycin).10 There are different motivations for patients to join a clinical trial, particularly for rare diseases; these include seeking a cure, improving available treatments, personal gain, or scientific curiosity. The key motivation of our patient to participate in clinical trials (3 so far) was altruism, helping to advance medical knowledge and the opportunity to improve the health of others.
Two observations are relevant to her clinical course in the era of choices.11 First, patients with GD often maintain stability even when they experience significant disease manifestations, whether they are untreated or nonresponders. Therefore, “maintaining stability” should not be considered a positive endpoint in future clinical trials.12 Second, massive focal lesions within an enlarged spleen predict a poor platelet and splenic response to ERT13 and probably to any specific therapy. Thus, these findings may represent a rare indication for splenectomy even nowadays, which can be performed by hand-assisted laparoscopy even for huge spleens.14
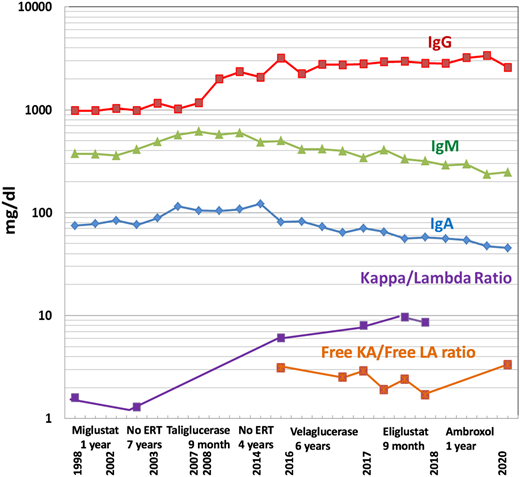
Immunoglobulins and free light chain ratios over time, showing increased IgG levels from 2013 with a gradual decrease of IgM and IgA and a stable free light chain ratio despite various GD-specific therapies.
Immunoglobulins and free light chain ratios over time, showing increased IgG levels from 2013 with a gradual decrease of IgM and IgA and a stable free light chain ratio despite various GD-specific therapies.
Case report part 4: GD and monoclonal gammopathy of undetermined significance
During the follow-up period, a gradual increase in quantitative serum immunoglobulin G (IgG) was noted but did not raise concerns, given the relatively high prevalence of polyclonal gammopathy in GD.15 However, in 2017, a monoclonal gammopathy IgGκ was confirmed by immunofixation, with a mildly elevated free κ/λ ratio of 3.1 and serum IgG of 27.9 mg/dL (Figure 3). The patient’s platelet count was 20 × 109/L, and her hemoglobin was 9.2 g/dL. Her renal function, serum albumin, lactate dehydrogenase, calcium, phosphorus, and C-reactive protein levels were within normal limits, and her β2-microglobulin concentration was 3.72 mg/L.
Trephine bone marrow biopsy showed massive infiltration by Gaucher cells; all lineages were present; and 10% infiltration by plasma cells expressing monoclonal κ-light chains was present. These findings were summarized by the pathologist as compatible with GD and multiple myeloma (Figure 4). Flow cytometry and positron emission tomography/computed tomography were nondiagnostic; the finding of a myeloma fluorescence in situ hybridization panel was negative; and the overall picture did not fulfill the diagnostic criteria for myeloma. Erythropoietin and zoledronic acid were added as supportive therapies, and eliglustat was added as GD-specific therapy. The patient received 9 months of eliglustat 84 mg twice daily (as a cytochrome P450 2D6 [CYP2D6] extensive metabolizer), again without effect. There was no significant change in the patient’s immunoglobulin profile thereafter.
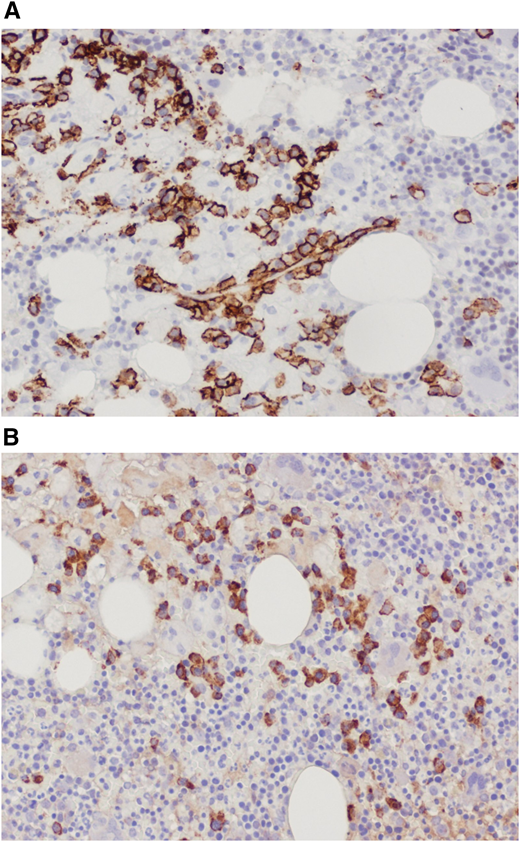
(A) Bone marrow trephine biopsy showing CD138 staining of plasma cells. (B) Bone marrow trephine biopsy showing κ-light chain staining.
(A) Bone marrow trephine biopsy showing CD138 staining of plasma cells. (B) Bone marrow trephine biopsy showing κ-light chain staining.
The association between GD and monoclonal gammopathy of undetermined significance (MGUS) was first reported in 1968. In the 1980s, it was suggested that the production of polyclonal and monoclonal gammopathies, including MGUS and myeloma, may be due to chronic stimulation of the immune system secondary to inflammatory cytokines, particularly interleukin-6, derived from the storage cells.16 Multiple myeloma is recognized as a GD-related comorbidity,17 and hence, immunoglobulins are included in the laboratory tests done during follow-up visits of our adult patients.18 Estimations of the prevalence of MGUS range between 1% and 39% of all adult patients with GD, and the magnitude of the actual myeloma risk is between 5- and 50-fold greater than in the general population.19 Recently, there has been a heated scientific debate regarding whether the monoclonal antibodies, and accordingly the underlying mechanism of GD-related myeloma, are specifically directed at macrophage CD1d-presented glycosphingolipid antigens20 or at saposin C.21
Although no therapy has been proved to ameliorate the paraproteinemia in GD, on the basis of anecdotal reports showing antimyeloma effects of eliglustat in a mouse model,20,22 eliglustat was attempted unsuccessfully in our patient. It is also unknown if early administration of any GD-specific therapy can prevent MGUS or myeloma or modify the clinical course or response to myeloma therapy in GD. Because glycosphingolipids may impact the immune system in opposing directions,23 it could be speculated that in some patients with GD, the accumulated glycosphingolipids may promote mainly immune tolerance and thereby paradoxically be associated with a milder course of disease or a lesser risk of progression from MGUS to myeloma.
Case report part 5: novel therapeutic approaches for GD
Currently, after another year of no GD-specific therapy, the patient joined investigator-initiated research evaluating high-dose ambroxol as a glucocerebrosidase PC (ClinicalTrials.gov identifier NCT03950050), so far with no response. The 12 months are going to end in the northern summer of 2020, and a decision needs to be made regarding the next step.
Although it is very unusual for a patient with GD type 1 to fail to respond to all conventional, registered therapies, consideration of alternative and novel modalities becomes relevant. The option of splenectomy has come up repeatedly because it is likely to end most of her key disease abnormalities. Although uncommon in the era of ERT/SRT, there are several cases of nonresponders or patients unable to receive ERT (mainly due to allergies or neutralizing antibodies) or SRT (CYP2D6 ultrarapid metabolizers)24 whose disease parameters and quality of life improved dramatically after splenectomy. With regard to quality of life, a greater emphasis is given nowadays to disease-specific patient-reported outcome measures, which are also required by regulatory agencies in the assessment of new drugs, including for rare diseases.25 In a first study of GD-specific patient-reported outcome in a relatively large cohort of adult patients, we have recently reported that despite the known association between GD and cancer, mainly myeloma, almost two-thirds of patients reported a lack of concern about this risk.26
The use of PC is based on the recognition that beyond a glycolipid storage disease, GD is also a protein-misfolding disorder,27 and the proof of concept was reported in 2013.28 Unfortunately, because ambroxol is inexpensive and exists in different generic formulations, a formal pharmaceutical company–sponsored clinical trial has not been performed. After encouraging data from IIR in Japan in patients with neuronopathic GD,29 we have launched an open-label clinical trial of ambroxol in patients with suboptimal response to ERT as well as for untreated patients with one of the following 3 abnormalities: thrombocytopenia, elevated LysoGb1, or reduced lumbar spine bone density. Our patient will soon complete the high-dose ambroxol IIR (600 mg/day), but again with no apparent improvement, so the next steps are under discussion.
Apart from splenectomy, there are several new clinical trials that are under consideration for our patient. First is arimoclomol, an investigational heat shock protein amplifier.30 Results from a 6-month phase 2 dose-finding study (NCT03746587) have just been announced, and when or if a phase 3 trial opens, it could be an interesting oral option (the preferable mode of administration for patients with GD).31 Second is gene therapy; there are currently 3 companies applying for phase 1/2 clinical trials for either ex vivo or in vivo GD trials. Although our patient might not fulfill the inclusion and exclusion criteria of the early trials (particularly due to her age), the various risks associated with these novel approaches are quite significant.
It seems that our patient will continue to refuse splenectomy, will not be enthusiastic or able to join the forthcoming gene therapy trials, and hence will remain untreated, as patients with GD did in the past, before the advent of specific therapies, and we hope that she will continue to maintain an uncomplicated stable status.
Acknowledgments
We thank Constantin Reinus of the Department of Pathology at Shaare Zedek Medical Center for preparation of the histological figures.
Correspondence
Ari Zimran, Shaare Zedek Medical Center, 12 Shmuel Bait St, PO Box 3235, Jerusalem 9103102, Israel; e-mail: azimran@gmail.com.
