

Abstract
Cellular-redirecting therapies, including bispecific T-cell engagers and chimeric antigen receptor (CAR) T cells, are rapidly changing the treatment landscape of hematologic malignancies and solid tumor malignancies. I will discuss the unique safety profile and logistical aspects that pose challenges and opportunities for the safe and successful delivery of these therapies. Close interaction, communication, and established partnerships between the primary oncologist, the disease specialist, and the immune effector cell provider will be needed to provide optimal care longitudinally for any patient. I will discuss practical ways for any program to deliver these therapies and how future advances may widen availability beyond just a few centers.
Learning Objectives
Identify best practices for safe and successful delivery of cellular redirecting therapies
Identify opportunities to anticipate and address acute phase and late phase toxicity
Prepare and plan for a clinical model that best fits your practice
Clinical case
A 52-year-old man with germinal center B-cell diffuse large B-cell lymphoma (DLBCL), without MYC translocation, is referred for consideration of chimeric antigen receptor (CAR) T therapy. The patient was diagnosed 2 years ago and underwent 6 cycles of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone, achieving complete remission (CR). One year later he relapsed and underwent salvage with 3 cycles of rituximab, ifosfamide, carboplatin, and etoposide followed by autologous hematopoietic cell transplantation with a carmustine, etoposide, cytosine arabinoside, and melphalan conditioning regimen. He achieved CR, but 6 months later he presents with relapse. Clinically he is doing well, and aside from hypertension and hypercholesterolemia he is healthy. He continues to perform activities of daily living without difficulty and has an excellent performance status. You agree he is a good candidate for CAR T therapy. Are you and your institution ready to proceed with this type of therapy? What are the next steps in shepherding this patient safely and successfully through the CAR T process?
Introduction
In 2017 the US Food and Drug Administration (FDA) approved blinatumomab, a CD19:CD3 bispecific T-cell engager, for the treatment of relapsed or refractory B-cell acute lymphoblastic leukemia, heralding a new era of immunotherapy.1 This approval was shortly followed by the approval of 2 CD19-directed CAR T cell products, tisagenlecleucel for the treatment of relapsed or refractory B cell acute lymphoblastic leukemia 2,3 and DLBCL and axicabtagene ciloleucel4 for the treatment of DLBCL. Since then the number of ongoing clinical trials and therapies in development is mind boggling. In 2020, the FDA predicted the development of >200 investigational new drugs per year and expected to approve 10 to 20 cell and gene therapy products per year by 2025.5 Furthermore, it is becoming clear that although there are common themes across the different therapies, individual products behave differently between disease types and even within diseases; some of these differences include significant variation in dosage levels, degree and duration of cytokine release syndrome (CRS) and neurotoxicity, variable duration of cytopenias, and variable time to onset of CRS. 6-15 These unique qualities may pose both a challenge and an opportunity for various delivery and access methods.
Practical aspects
Autologous CAR T process
Without a doubt, the resource-heavy processing and delivery of autologous CAR T products present unique challenges. A pictorial representation of the basic autologous CAR T process is shown in Figure 1. Initially the process mirrored the one already established by transplant programs. But even then, the processes in place needed adjustments, and specific immune effector cell (IEC) programs and paradigms were created. Figure 2 depicts some of the practical and logistic differences between the standard autologous hematopoietic cell transplantation process and the autologous CAR T process. Although the discussion that follows is aimed at the delivery of autologous CAR Ts, the development of off-the-shelf cellular and antibody-based products may allow simplification of at least the initial processes.
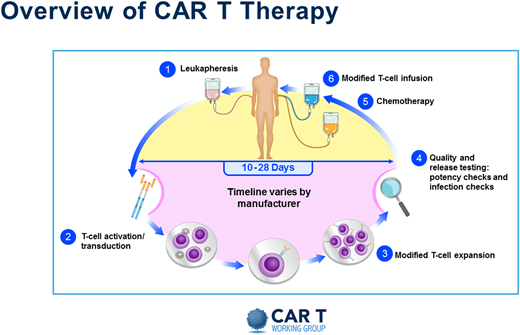
Overview of CAR T therapy. Reproduced with permission from the Slide Library of the CAR T Working Group (v2 9.4.2019).
Overview of CAR T therapy. Reproduced with permission from the Slide Library of the CAR T Working Group (v2 9.4.2019).
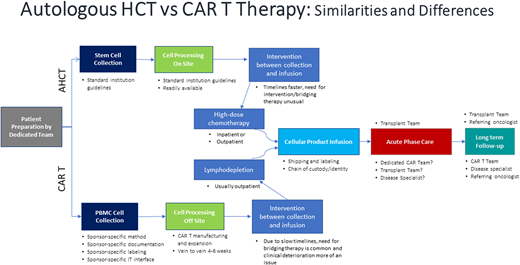
Autologous hematopoietic cell transplantation versus CAR T therapy: similarities and differences.
Autologous hematopoietic cell transplantation versus CAR T therapy: similarities and differences.
Toxicity profile
The toxicity profiles associated with cellular redirecting therapies have been extensively described elsewhere and are summarized in Table 1.6-9 In general, toxicity can be divided into an acute phase (30 days) and a late phase (beyond 30 days). In the acute phase, CRS, neurotoxicity, and cytopenias predominate. The rapidity and onset of these potential toxicities require multidisciplinary expert vigilance, availability, and management. These are treated mostly in the inpatient setting but are slowly starting to be managed on an outpatient basis for some patients.
CAR T–associated toxicities
| Acute phase (D0-30) | Late phase (D30+) |
|---|---|
| • CRS | • Persistent cytopenias |
| • Immune effector cell–associated neurotoxicity syndrome | • B-cell aplasia and hypogammaglobulinemia |
| • Cytopenias | • ?IVIG replacement |
| • Macrophage activation syndrome or hemophagocytic lymphohistiocytosis, is a very rare and severe form | • T-cell deficiency |
| • Disseminated intravascular coagulopathy | • Pneumocystis jirovecii pneumonia and varicella-zoster virus prophylaxis, other? |
| • Infection prophylaxis | |
| • B-cell aplasia and hypogammaglobulinemia | • Residual effects of acute toxicity |
| • Life threatening if not managed by expert multidisciplinary team | • Delayed CRS and neurotoxicity is rare but can occur. |
| • Tumor lysis is rare and probably varies by disease and disease burden. | • Impaired quality of life: fatigue, memory issues, not yet well described |
| Acute phase (D0-30) | Late phase (D30+) |
|---|---|
| • CRS | • Persistent cytopenias |
| • Immune effector cell–associated neurotoxicity syndrome | • B-cell aplasia and hypogammaglobulinemia |
| • Cytopenias | • ?IVIG replacement |
| • Macrophage activation syndrome or hemophagocytic lymphohistiocytosis, is a very rare and severe form | • T-cell deficiency |
| • Disseminated intravascular coagulopathy | • Pneumocystis jirovecii pneumonia and varicella-zoster virus prophylaxis, other? |
| • Infection prophylaxis | |
| • B-cell aplasia and hypogammaglobulinemia | • Residual effects of acute toxicity |
| • Life threatening if not managed by expert multidisciplinary team | • Delayed CRS and neurotoxicity is rare but can occur. |
| • Tumor lysis is rare and probably varies by disease and disease burden. | • Impaired quality of life: fatigue, memory issues, not yet well described |
Late phase toxicity for the most part is dominated by persistent or recurrent cytopenias and increased infection risk. Issues pertaining to infectious prophylaxis, revaccination, immunoglobulin deficiency, and need for replacement predominate. Longer-term sequelae such as fatigue, memory loss, and lapses in concentration are becoming better described.9,12,13 Although the optimal management continues to evolve and be defined, clear processes and resources are needed for the safe delivery of these therapies.
Requirements of an IEC program
Perhaps the best way to identify the practical needs for a new program is to address the steps that a patient undergoing CAR T therapy will traverse. Figure 3 breaks this process into 8 different steps.
The first is intake and triage, initiated by the patient or by a referring provider. Ideally, it involves a dedicated triage nurse or navigator, a dedicated CAR T coordinator, and an insurance benefits or financial coordinator.
This first step leads to a consultation with an IEC provider, who will determine eligibility. Given the current indications of the FDA-approved products in the relapsed or refractory setting of aggressive diseases, moving forward quickly is important. If the patient is found eligible, the CAR T coordinator then sets in motion all necessary procedures and communications with all appropriate parties, including apheresis staff assessment to determine the type of catheter needed, usually large-bore peripheral IV versus a temporary trifusion catheter, and to secure the apheresis date and time; and social work evaluation to assess for any concerns about compliance, caregiver support, financial resources, and ability to secure local housing if needed.
The next step is the collection, processing, and shipping or manufacturing of cells. The vein-to-vein process will vary, but anticipate on average 4 to 6 weeks. Once the CAR T product has been manufactured, it is shipped and received by the cell processing laboratory in preparation for infusion. A thorough analysis of expected volume increase and additional infrastructure needs must be undertaken to anticipate space, equipment, and staffing needs.
After apheresis and during the manufacture of the CAR T product, many patients will need bridging therapy. The goal of bridging therapy is to control the patient’s disease but not induce toxicity that may make the patient ineligible to receive the CAR T product. This therapy can be delivered by the IEC provider or the referring oncologist or disease specialist.
Once the product is ready for infusion, the patient is reevaluated by the IEC provider to ensure that no new problems have arisen that would make the patient ineligible to proceed as planned. Currently, most CAR T products are indicated in aggressive tumors in the relapsed and refractory setting. Therefore, deterioration of performance status and organ function and concurrent infections can often halt the process. It is important that these are resolved before treatment begins. If the patient is found eligible, the team (IEC provider, outpatient nursing, and pharmacy) prepare and proceed with lymphodepleting chemotherapy, usually delivered over 3 days. This is often done on an outpatient basis but can be done on an inpatient basis if logistically more feasible; the inpatient setting is the most common. The patient then undergoes infusion of the product, usually 2 to 7 days after completion of lymphodepletion.
After infusion, the patient is considered to be in the acute phase of their potential toxicity. This phase usually lasts 30 days. With current products, the first 2 weeks are the highest risk for development of CRS, neurotoxicity, and pancytopenia. Careful evaluation, coordination, observation, and treatment are carried out by the IEC provider in conjunction with the medical intensive care unit, the emergency room team, neurology, and other consultants as needed. Of course, experienced nurses and pharmacists are paramount. Ensuring emergent availability of blood products and anticytokine drugs such as tocilizumab and anakinra is mandatory.
At day 30, if clinically well, the patient is released to the referring provider, and long-term surveillance begins. This is usually done by the referring oncologist in consultation with the IEC provider. During this time, patients are in their late phase of potential toxicity and often still cytopenic, and they may need transfusion and growth factor support. Patients continue to have both humoral and cellular immunosuppression. Although the exact antimicrobial prophylaxis has not been standardized, most experienced centers recommend prophylaxis for at least varicella-zoster virus and Pneumocystis jirovecii pneumonia and possibly intravenous immunoglobulin (IVIG) infusion. A plan must be put in place to ensure the safety of these patients. Frequent communication between the primary oncologist and the IEC provider is paramount.
Finally, continuous reporting of outcomes is currently required by the Centers for Medicare and Medicaid (CMS). This can be accomplished in various ways; one is to use the established Centers for International Blood and Marrow Transplant Research reporting already being done by many centers.
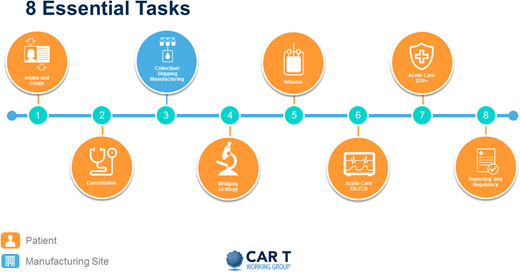
Illustration of a patient’s journey through the CAR T process. Reproduced with permission from the Slide Library of the CAR T Working Group (v2 9.4.2019).
Illustration of a patient’s journey through the CAR T process. Reproduced with permission from the Slide Library of the CAR T Working Group (v2 9.4.2019).
Regulatory hurdles
CMS has established a risk evaluation and mitigation strategy (REMS) program to define criteria for a center to qualify as a provider of FDA-approved IECs and be reimbursed (https://www.cms.gov/). REMS is a safety program for medications with serious safety concerns. It is designed to reinforce safe medication use and applies only to commercial, licensed products and not research products. It is the responsibility of the manufacturer to develop the REMS program based on FDA guidelines and to implement and monitor compliance. It is the responsibility of the IEC program or provider to enroll and become certified in the REMS program and comply with REMS regarding training, consenting, prescribing, patient management, and compliance. This includes all staff who will be involved in the care of these patients. Centers can usually meet these requirements by being accredited by the Foundation for Accreditation of Cellular Therapies or Joint Accreditation Committee, International Society for Cell & Gene Therapy/European Society for Blood and Marrow Transplantation, although this is not necessary (http://www.factwebsite.org/). Each center must establish standard operating procedures that conform and meet all the REMS requirements. CMS has also established a requirement to provide continuous long-term outcome data under the coverage with evidence development program (https://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development). Finally, a thorough understanding and assessment of the reimbursement process are necessary before a center decides to become a provider for CAR T therapy. The financial considerations are complex, continue to evolve, and can lead to significant unreimbursed costs of care. A description of these requirements is beyond the scope of this discussion but is well documented, and centers are urged to review them for guidance before moving forward in establishing a new IEC program.
Building a team and best practices from experienced centers
It is imperative to develop a core team of providers dedicated to IEC therapy. For some centers, this will be a new, independent team or clinical unit; for others the existing transplant team will absorb this task; and for others it may be a hybrid entity where resources are shared between the IEC team and the transplant program. Each center will determine which model makes the most sense for them. Regardless of the underlying structure, the IEC team should be multidisciplinary and include the IEC provider, CAR navigators, dedicated intensive care physicians, neurologists and other consultants, pharmacy, and collection and processing facilities. Ideally, ongoing education and communication should occur among the various stakeholders. Each program should develop and disseminate written guidance for management of complications, including the use of anticytokine therapy and corticosteroids.
Communication between all providers is imperative. All involved providers should be continuously educated on all aspects of IEC therapy. Multidisciplinary rounds, weekly grand rounds, or similar rounds are encouraged. Standard operating procedures should be developed to facilitate communication for after-hours providers. Ensure rapid availability of ICU support, neurology, infectious disease, and other consultants as needed. The ICU should be aware of all CAR T recipients admitted to the IEC service.
Nurses should be well trained and dedicated to the care of CAR T recipients. Nurses should undergo continuous education in CAR T therapy and adverse event management. Incorporating a symptom checklist and neurologic assessment at least once per shift often reinforces the need for continued surveillance and early detection of symptoms.
Pharmacy should be aware of the plans for lymphodepletion and CAR T infusion. The pharmacist should provide supervision to ensure proper management of CAR T therapy side effects and ensure that anticytokine therapy (ie, tocilizumab) is available for use emergently, ideally infusion within 1 hour of the order being placed.
Emergency room staff should be educated about the specific needs of this patient population. They should have access and knowledge of CRS and neurotoxicity management algorithms, and they should promptly communicate with the IEC team and medical intensive care unit if needed. Ideally, they should attend multidisciplinary team meetings.
Perhaps the most important measure is embracing the patient and caregiver as integral members of the care team. This is especially true in the outpatient setting, but even in the inpatient setting, educating patients and caregivers will lead to prompt detection of CRS, neurotoxicity, and other side effects. Early identification of symptoms can significantly improve intervention and hopefully decrease high-grade toxicity and poor outcomes.
Although these recommendations focus mostly on the acute phase of management, the paradigm applies to the late phase management as well. Although unusual, delayed CRS and neurotoxicity could occur. Infections become a higher risk during this time. At this point the referring oncologist becomes the primary caregiver, and education and communication are of the utmost importance. Best practices for long-term management after CAR T therapy are still evolving. However, standards of care and practice must be developed for any center, and all members of the team, including the referring providers, must be familiar with these recommendations. Written guidelines for the use of prophylactic antimicrobials, use of growth factors, use of IVIG replacement, revaccination strategies, and disease monitoring and restaging are extremely helpful to minimize errors of omission. A long-term coordinator could be the liaison between the primary oncologist and the IEC team and can ensure proper communication and implementation of the various long-term needs for the patient.
Future
For now, most FDA-approved therapies require a period of inpatient treatment. However, it appears there may be certain intrapatient variables such as burden of disease, high inflammatory markers, comorbidities that may predict higher risk of high-grade toxicity.14-16 Similarly, the disease being treated, the specific target (CD19 vs BCMA), the cell modified (T cell vs natural killer cell), and other characteristics may continue to challenge the need for inpatient administration and supervision.10,11,17-20 Currently, most products are autologous; they not only require particular resources but also can make the coordination and prompt delivery of therapy cumbersome. Off-the-shelf cellular and antibody-based products are being evaluated in clinical trials.1,21,22 Theoretically these therapies could simplify the treatment algorithm. Furthermore, as these therapies continue to expand beyond the current disease indications and eventually solid tumors,19 with the ability to predict and mitigate toxicity, these therapies will become more and more amenable to outpatient dosing for many patients if not the majority. Therefore, the facilities that will be able to deliver these therapies are likely to expand beyond the current structure. In anticipation of this potential, various groups such as the Community Oncology Alliance and the Association of Community Cancer Centers have been active in establishing educational resources, reviewing requirements, and providing guidance in preparation for possible incorporation of these therapies. Each individual program will need to determine the approach that makes best use of its resources.
Conclusion
As IEC therapy continues to expand, it is likely that current providers will need to adjust, and more facilities will need to become providers of these therapies. It is important to realize that establishing an IEC program requires very specific resources and structures to allow the safe delivery of these therapies. A careful assessment of an individual program’s capabilities and potential need for partnership is imperative. Each individual program will need to determine the best approach that makes best use of resources. Paramount will be communication between all involved parties, including referring providers, disease specialists, IEC providers, and consultants.
Conflict-of-interest disclosure
The authors’ institution receives research funds under J.G.B.’s name from the following: Abbvie, Amgen, Acetylon, Bluebird, BMS, Celgene, Celularity, Constellation, CRISP Therapeutics, CURIS, EMD Sorono, Genentech, Glenmark, Ichnos, Janssen, Kesios, Lilly, Novartis, Poseida, Sanofi, Takeda, Teva, and Vivolux. Additionally, the institution received consultancy payments from the following: Amgen, BioClinica, Bluebird, BMS, Celgene, CRISPR Therapeutics, Janssen, Karyopharm, Kite Pharma, Legend, Prothena, SecuraBio, Servier, and Takeda.
