

Abstract
Myelodysplastic syndromes (MDS) are characterized by heterogeneous biological and clinical characteristics, leading to variable outcomes. The availability of sophisticated platforms of genome sequencing allowed the discovery of recurrently mutated genes, which have led to a new era in MDS. This is reflected by the 2016 update of the World Health Organization classification, in which the criteria to define MDS with ringed sideroblasts include the presence of SF3B1 mutations. Further, the detection of somatic mutations in myeloid genes at high variant allele frequency guides the diagnostic algorithm in cases with cytopenias, unclear dysplastic changes, and normal karyotypes, supporting MDS over alternative diagnoses. SF3B1 mutations have been shown to play a positive prognostic role, while mutations in ASXL1, EZH2, RUNX1, and TP53 have been associated with a dismal prognosis. This is particularly relevant in lower- and intermediate-risk disease, in which a higher number of mutations and/or the presence of “unfavorable” somatic mutations may support the use of disease-modifying treatments. In the near future, the incorporation of mutation profiles in currently used prognostication systems, also taking into consideration the classical patient clinical variables (including age and comorbidities), will support a more precise disease stratification, eg, the assignment to targeted treatment approaches or to allogeneic stem cell transplantation in younger patients.
Learning Objectives
Discuss the impact of somatic mutations in MDS and how to integrate molecular data within current diagnostic and prognostic classifications
Review the clinical management of patients with MDS according to the disease mutational status and identify actionable targets
CLINICAL CASE 1
A 78-year-old woman with an unremarkable medical history presented to our clinic with isolated macrocytic anemia (hemoglobin [Hb], 9 g/dL; mean corpuscular volume, 107 fL). Metabolic as well as nutritional deficiencies and other common causes of anemia were ruled out after an initial diagnostic workup, while the endogenous erythropoietin (EPO) level was 358 mU/mL. The bone marrow (BM) evaluation revealed expansion of erythropoiesis (70% of marrow cellularity) with marked dysplastic changes in 20% of erythroid cells and the presence of 1% blasts and 12% ring sideroblasts (RS) by iron staining (Perls' Prussian blue; Figure 1). The karyotype was normal, while next-generation sequencing (NGS) mutational analysis identified a canonical SF3B1K700E mutation at a variant allelic frequency (VAF) of 21%. Based on these findings, a myelodysplastic syndrome (MDS) with RS and single-lineage dysplasia was diagnosed.

Perls' Prussian blue stain of the patient's BM smear showing RS. Classical type 3 RS are shown (according to Mufti et al57 ), with multiple siderotic granules in a perinuclear position surrounding the nucleus (lower-right cell) or encompassing at least one-third of the nuclear circumference (middle cell).
Perls' Prussian blue stain of the patient's BM smear showing RS. Classical type 3 RS are shown (according to Mufti et al57 ), with multiple siderotic granules in a perinuclear position surrounding the nucleus (lower-right cell) or encompassing at least one-third of the nuclear circumference (middle cell).
Introduction
MDS is a highly heterogeneous group of disorders defined by the presence of ineffective hematopoiesis with peripheral blood (PB) cytopenias, dysplastic changes in ≥10% of cells of one or more myeloid lineages, and a variable risk of progression to acute myeloid leukemia (AML).1 Clinical presentations of MDS reflect its biological heterogeneity. Based on the major risk-prognostication systems, the International Prognostic Scoring System (IPSS) and its revision (IPSS-R),2,3 two-thirds of patients will present with lower-risk disease (LR-MDS) and are generally treated with supportive care. In these cases the goal of treatment is the amelioration of cytopenias, represented by anemia in about 70% of cases, through transfusion support, erythropoietin administration, or lenalidomide treatment in MDS with del(5q). Conversely, higher-risk MDS patients, defined by intermediate to very high IPSS-R,4 require disease-modifying treatments (DMT) such as hypomethylating agents (HMA) and a timely evaluation for allogeneic hematopoietic stem cell transplantation (HSCT), the most effective treatment and the only potentially curative option.5
Integration of molecular data into classification and prognostication systems
Paralleling the variable clinical presentation, MDS biology is fascinatingly complex. In the last decade, the availability of sophisticated genome-sequencing platforms led to the identification of recurrently mutated genes, shedding light on new aspects of the disease pathobiology. Accordingly, del(5q) and SF3B1 mutations have been incorporated in the most recent World Health Organization (WHO) classification and define specific MDS subtypes.1 However, the common risk-prognostication tools (ie, the IPSS and IPSS-R), used worldwide for clinical decision-making and trial eligibility, consider only clinical/cytogenetic variables. Briefly, the IPSS score groups patients in a 4-tier model (low, intermediate-1, intermediate-2, and high),2 while the more recent IPSS-R includes 5 risk groups (very low, low, intermediate, high, and very high), improving stratification of karyotype and of the proportion of blasts and taking into account the severity of cytopenias (Table 1).3 The IPSS-R, initially developed in patients receiving supportive treatment, has since been validated in patients treated with DMT and therapy-related MDS.6,7
Comparison of existing prognostication models in MDS
| Model | Variables | C index | Reference |
|---|---|---|---|
| International Prognostic Scoring System (IPSS)2 | Karyotype BM blasts % Number of cytopenias | 0.65 | 2 |
| Revised International Prognostic Scoring System (IPSS-R)3 | Karyotype BM blasts % Degree of cytopenias | 0.67 | 3 |
| MD Anderson Cancer Center (MDACC)55 | Karyotype BM blasts % Degree of cytopenias Age Performance status Prior transfusion | 0.65 | 49 |
| World Health Organization-based Prognostic Scoring System (WPSS)56 | Karyotype WHO category Transfusion requirement | 0.65 | 50 |
| Genoclinical model according to Haferlach et al8 | Gender Age Degree of cytopenias (PLT, Hb) BM blasts % Karyotype ASXL1, CBL, ETV6, EZH2, KRAS, LAMB4, NCOR2, NF1, NPM1, NRAS, PRPF8, RUNX1, TET2, TP53 | NA | 8 |
| Genoclinical model according to Nazha et al10 | Karyotype BM blasts % Degree of cytopenias Age WHO category Disease ontogeny (secondary vs de novo) TP53, EZH2, and SF3B1 | 0.71 | 10 |
| Genoclinical model according to Bersanelli et al15 | 63 clinical and molecular variables | 0.75 | 14 |
| Model | Variables | C index | Reference |
|---|---|---|---|
| International Prognostic Scoring System (IPSS)2 | Karyotype BM blasts % Number of cytopenias | 0.65 | 2 |
| Revised International Prognostic Scoring System (IPSS-R)3 | Karyotype BM blasts % Degree of cytopenias | 0.67 | 3 |
| MD Anderson Cancer Center (MDACC)55 | Karyotype BM blasts % Degree of cytopenias Age Performance status Prior transfusion | 0.65 | 49 |
| World Health Organization-based Prognostic Scoring System (WPSS)56 | Karyotype WHO category Transfusion requirement | 0.65 | 50 |
| Genoclinical model according to Haferlach et al8 | Gender Age Degree of cytopenias (PLT, Hb) BM blasts % Karyotype ASXL1, CBL, ETV6, EZH2, KRAS, LAMB4, NCOR2, NF1, NPM1, NRAS, PRPF8, RUNX1, TET2, TP53 | NA | 8 |
| Genoclinical model according to Nazha et al10 | Karyotype BM blasts % Degree of cytopenias Age WHO category Disease ontogeny (secondary vs de novo) TP53, EZH2, and SF3B1 | 0.71 | 10 |
| Genoclinical model according to Bersanelli et al15 | 63 clinical and molecular variables | 0.75 | 14 |
PLT, platelets; NA, not available.
The advent of NGS and the growing body of evidence concerning the prognostic impact of somatic mutations in MDS have posed new challenges, such as the incorporation of this information into the established risk-stratification models. To this end, several studies have demonstrated the impact of somatic myeloid gene mutations on survival in patients with MDS.8-10 The number of mutations itself has been shown to play a significant prognostic role by most studies, with patients carriers of over 3 driver mutations characterized by reduced survival and a higher probability of leukemia progression.11-13 This is particularly relevant in lower- or intermediate-risk MDS, in which a higher number of somatic mutations, or the presence of “unfavorable” mutations (ie, TP53), may refine the prognosis and set the indication for DMT or HSCT.14 In a survey of 104 genes, Haferlach et al showed by univariate analysis that among the 25 genes found to affect survival, only 5 (ASXL1, KRAS, PRPF8, RUNX1, and SF3B1) retained significance after correcting for confounding factors.8 These mutations were incorporated into a novel hybrid genomic-clinical model that more accurately predicted MDS patients' prognosis when compared to both the IPSS-R and the “gene-only” model.8 Another study from the Cleveland Clinic identified one favorable (SF3B1) and 5 unfavorable gene mutations (ASXL1, EZH2, NPM1, RUNX1, and TP53) in a cohort of 508 MDS patients.10 These data led to the design of a score, including age, IPSS-R, and the mutational status of EZH2, SF3B1, and TP53, that outperformed the classical MDS risk-prediction models (Table 1) and could predict longitudinal disease evolution regardless of treatment.10 Of note, these scores unveiled discrepancies in risk assignment for a not negligible fraction of patients. For instance, if taking into consideration the IPSS-R, the hybrid “powered” model upstaged 58% of patients previously categorized as intermediate risk to high as well as 26% from lower to high risk.9
More recently, by applying Bayesian networks and Dirichlet processes in a study enrolling 2043 patients annotated for the presence of mutations in 47 frequently mutated genes, Bersanelli et al identified 8 clusters, each characterized by different genetic lesions, clinical phenotypes, and prognoses.15 By using a random-effects Cox multistate model, the authors combined 63 clinical and genomic variables to profile individual patients' prognoses, yielding a C-index of 0.75 and 0.74 when cross-validated internally and in an independent cohort, respectively.15 This seminal study paves the way for an MDS prognostication system not only accounting for traditional clinical and morphological variables but also incorporating recent advances in MDS biology.
Limitations to the application of genomics to MDS diagnostics
If the idea of a hybrid genomic-clinical model sounds appealing, several factors still play a role in determining the actual feasibility and reproducibility of such an approach. There is no general consensus yet on sequencing techniques and data analysis across laboratories. The clinical impact of some variants on patients' outcomes remains unknown, and of utmost importance, biological differences in mutation characteristics such as VAF, type (missense vs truncating), and location (functional domain vs other positions across the gene) are still not homogeneously considered. For instance, TP53 mutations are classically considered prognostically unfavorable. However, a recent cooperative study demonstrated that their clinical impact is beyond the mere binary presence/absence.16 Indeed, Bernard et al confirmed that besides VAF, the TP53 allelic state is a strong predictive factor, showing that patients with monoallelic TP53 mutations and VAF ≤22% were characterized by survival and response to therapy similar to wild-type counterparts.16
In addition, the costs of genome-scanning approaches (despite dropping dramatically in the last years) still represent an obstacle for a worldwide hybrid genomic-clinical application used on a day-by-day basis, especially in developing countries.
CLINICAL CASE 1 (continued)
Our 78-year-old woman had an LR-MDS (IPSS score, 0; IPSS-R, 2). According to the common guidelines, the patient was started on alpha EPO (40 000 UI subcutaneously once weekly, increased to 80 000 UI weekly after 12 weeks). However, after 6 months the hemoglobin levels remained unchanged, and she eventually became transfusion dependent (1 red blood cell unit [RBC]/week). At this point, considering the diagnosis of SF3B1-mutant MDS with RS and single-lineage dysplasia and the erythropoiesis-stimulating agent failure, the patient was enrolled in the MEDALIST trial and started on luspatercept.17 A progressive increase in hemoglobin levels and a reduction of transfusion needs were observed after 8 weeks, with achievement of a minor hematologic improvement—erythroid response (1 RBC unit/3 weeks), according to the revised International Working Group 2018 hematological response criteria— still persisting after 27 months on treatment.18
Key clinical point
In Case 1, the results of NGS were determinant in assigning the patient to the correct WHO-defined category (MDS-RS), in defining the prognosis, and in guiding the clinical management toward targeted treatment with luspatercept.
From biology to classification and tailored treatment: the case of SF3B1 mutations
As showcased by our patient, SF3B1-mutant MDS is an emblem of genotype/phenotype association, characterized by a specific prognostic outcome and the availability of a tailored treatment. SF3B1 is the most commonly mutated splicing-factor gene and defines a well-characterized group of MDS with RS, leading to its definition as “the lord of the rings” of MDS.19-21 The association is so strong that if the mutation is present, the threshold of RS proportion in the BM to satisfy the WHO MDS-RS criteria lowers from 15% to 5% (as in our patient).1 In a recent study, the International Working Group argues for the recognition of SF3B1-mutant MDS as a distinct nosologic entity and proposes specific diagnostic criteria (Table 2).22 This plea is supported by the strong genotype/phenotype association characterized by the prevalence of female sex, a higher degree of anemia and lower BM blast percentages, a favorable disease course, the presence of SF3B1 mutations as a founding event in the majority of cases, and the recent availability of a specific treatment.17,22
Proposed diagnostic criteria for the subentity of MDS with mutated SF3B1
| 1. Cytopenia defined by standard hematologic reference values |
| 2. Presence of a somatic SF3B1 mutation |
| 3. Isolated erythroid or multilineage dysplasia, with or without RS |
| 4. BM blasts <5% and PB blasts <1% |
| 5. WHO criteria not fulfilling any other category |
| 6. Normal karyotype or any cytogenetic abnormality other than del(5q), monosomy 7, inv(3) or abnormal 3q26, complex (≥3) |
| 7. Presence of any additional somatic gene mutation other than RUNX1 and/or EZH2* |
| 1. Cytopenia defined by standard hematologic reference values |
| 2. Presence of a somatic SF3B1 mutation |
| 3. Isolated erythroid or multilineage dysplasia, with or without RS |
| 4. BM blasts <5% and PB blasts <1% |
| 5. WHO criteria not fulfilling any other category |
| 6. Normal karyotype or any cytogenetic abnormality other than del(5q), monosomy 7, inv(3) or abnormal 3q26, complex (≥3) |
| 7. Presence of any additional somatic gene mutation other than RUNX1 and/or EZH2* |
Additional JAK2V617F, CALR, or MPL mutations strongly support the diagnosis of myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T). Adapted from Malcovati et al.22
SF3B1, together with del(5q), represents an ideal exemplum of how progresses in disease pathobiology can not only improve disease classification but also open possibilities for tailored treatments. The haploinsufficiency of CSNK1A1 and RPS14 represents the molecular lesions underlying the exquisite sensitivity of del(5q) cases to lenalidomide.23,24 The presence of SF3B1 mutations generates aberrant splicing events due to misrecognition of 3′ splice site with degradation of about 50% of the aberrant mRNAs via nonsense-mediated mRNA decay.25 Moreover, the ABCB7 and PPOX genes, involved in mitochondrial iron metabolism, have been found to be significantly downregulated in SF3B1-mutant MDS and are thought to be associated with the presence of RS.22,26 These molecular disturbances generate a higher degree of ineffective erythropoiesis and, therefore, of anemia. Luspatercept is a recombinant inhibitor of transforming growth factor β able to reduce SMAD2/3 signaling, overcoming the impairment imprinted by the genetic lesion and enabling late-stage erythroid differentiation. In the single-arm phase 2 trial (PACE-MDS) enrolling anemic LR-MDS patients, luspatercept induced hematologic improvement in 69% of RS-positive cases vs 43% of RS-negative cases. When looking at SF3B1 status, hematologic improvement was enriched in mutated patients (77% vs 40% in negative cases).27 These results were later confirmed in patients with MDS-RS by the randomized phase 3 MEDALIST trial, which showed that luspatercept (given at a dose of 1-1.75 mg/kg subcutaneously every 21 days) induced transfusion independence in 38% of patients at 8 weeks vs 13% in the placebo arm.17 This trial led to the approval of luspatercept by the US Food and Drug Administration and the European Medicines Agency for patients with LR-MDS with RS and transfusion-dependent anemia, who have an unsatisfactory response to or are ineligible for erythropoietin-based therapy.
CLINICAL CASE 2
A 55-year-old woman was admitted to our hospital after noticing the appearance of petechiae over her arms and legs and after a finding of pancytopenia (Hb, 9 g/dL; absolute neutrophil count, 0.8 × 109/L; platelets, 13 109/L) at a complete blood count evaluation. Her BM aspiration was hypocellular, and the karyotype was normal. A core biopsy confirmed the BM hypocellularity and showed <5% CD34+ cells with the presence of rare micromegakaryocytes and a normal reticulin-staining pattern. The patient was started on EPO 40 000 UI/week, but no changes in Hb levels were observed. NGS analysis using a 30-gene myeloid panel identified somatic mutations in 4 genes (DNMT3A, RUNX1, SRSF2, and TET2) at a median VAF of 12% (range, 1.5%-12.6%). Based on the presence of severe multilinear cytopenias and unilinear dysplasia and the evidence of <5% BM blasts, a diagnosis of MDS unclassifiable (MDS-U) with single-lineage dysplasia was made. The patient's IPSS score was 0.5 (intermediate-1), while IPSS-R was 4 (intermediate). Taking into consideration the young age at presentation, the severity of cytopenias leading to transfusion dependency of RBCs and platelets, and the mutational profile, a decision to proceed to upfront HSCT was made. She had no major complications from the transplantation procedure and is presently in complete remission (CR) at 24 months from HSCT.
Myeloid gene mutation analysis as a helpful diagnostic tool for MDS
As shown by the patient's presentation, the number and type of mutational events identified by NGS were helpful not only in supporting the diagnosis of MDS but also for deciding to proceed to HSCT. As mentioned before, somatic mutations (except SF3B1) are not part of current classification schemes of MDS, whose hallmark remains the presence of BM dysplasia, and there is no single mutation that can be considered 100% pathognomonic of the disease. However, the assessment of dysplasia in hypocellular cases with low blast counts and normal karyotypes poses challenges even well-experienced morphologists. In these cases, NGS may add important information since over 90% of patients with myeloid diseases have been shown to present somatic mutations.11,12 Indeed, when using a well-constructed gene panel, the absence of any molecular lesion in a patient with isolated cytopenia without BM dysplasia and a normal karyotype has a high negative predictive value and should prompt the consideration of alternative diagnoses (eg, autoimmune cytopenias), especially in younger patients.28 Conversely, the identification of somatic mutations may underpin a diagnosis of MDS, as in our patient, or direct physicians toward alternative diagnoses (ie, large granular lymphocytic leukemia in the case of STAT3 mutations).29 When applying NGS to routine diagnostics, clonal hematopoiesis of indetermined potential (CHIP) must be taken into account, since it is a very frequent finding in older adult individuals (up to 60% over the age of 80) and is characterized by mutations usually affecting one isolated gene (frequently, DNMT3A, TET2, and ASXL1), at a 2% to 30% VAF.14,30-32 In the context of CHIP, a high number of mutations and increased allelic burden are indications for frequent hematological follow-up since these mutations may be the prodromes for the onset of MDS.14,30,31 It is worthwhile to underline here that the presence of cytopenias and BM dysplasia remains an essential requirement for the diagnosis of MDS, while CHIP refers to a condition in which only somatic mutational events are present.1 Other four-letters acronyms are used to define the case of patients with somatic mutations and cytopenias in the absence of BM dysplasia (CCUS, clonal cytopenia of uncertain significance), BM dysplasia without cytopenia and clonal events (IDUS, idiopathic dysplasia of unknown significance), and cytopenia without dysplasia and mutations (ICUS, idiopathic cytopenias of uncertain significance).14
The increasing number of somatic mutations also mirrors disease progression, which is an intrinsic characteristic of MDS (Figure 2). This implies not only linear and branching clonal evolution, defined by the acquisition or loss of somatic mutations, respectively, but also increased DNA methylation levels.33,34 For these reasons, reassessment of the mutational profile at the time of disease progression is indicated to ensure treatment optimization, especially in younger patients.

Model of disease evolution in MDS.Upper panel: risk factors commonly associated with the development of myeloid disorders (germline variants, smoking, aging, and cancer treatment exposure such as chemo/radiotherapy). Middle panel: a hypothetical model of clonal evolution in which the founding event (or germline predisposition lesion) leads to subsequent acquisition/loss of new somatic mutations in a linear/branching fashion. Lower panel: the typical acquisition of methylation during MDS progression, which leads to the silencing of genes such as tumor suppressor and the disruption of many cell pathways (DNA repair, apoptosis, cell cycle, cell adherence).
Model of disease evolution in MDS.Upper panel: risk factors commonly associated with the development of myeloid disorders (germline variants, smoking, aging, and cancer treatment exposure such as chemo/radiotherapy). Middle panel: a hypothetical model of clonal evolution in which the founding event (or germline predisposition lesion) leads to subsequent acquisition/loss of new somatic mutations in a linear/branching fashion. Lower panel: the typical acquisition of methylation during MDS progression, which leads to the silencing of genes such as tumor suppressor and the disruption of many cell pathways (DNA repair, apoptosis, cell cycle, cell adherence).
Key clinical points
In Case 2 the decision to proceed to HSCT upfront was mostly based on clinical considerations, including the PB trilinear cytopenia leading to RBC and platelet transfusion dependency. The availability of the mutation profile, which showed the presence of 4 mutations also affecting RUNX1 and SRSF2 genes, was further supportive of the diagnosis of MDS and of the high probability of rapid disease progression.
Considerations on allogeneic HSCT in patients with MDS in the molecular era
Although allogeneic HSCT represents the only curative option in MDS, only 10% to 15% of patients will eventually undergo this procedure. Indeed, the demographics of the disease (ie, most patients are diagnosed with MDS at ages >70) constitute a major factor limiting HSCT accessibility. However, the risk of relapse remains the major outcome determinant even after HSCT. In this context, Della Porta et al identified lesions in ASXL1, RUNX1, and TP53 genes as independent predictors of dismal outcome.35 More importantly, the role of these somatic events was independent of the IPSS-R risk score, emphasizing once more the need to integrate clinical and molecular factors. Indeed, the combination of IPSS-R plus mutational characteristics changed the prediction of posttransplant outcomes for 34% of cases. Further, in another study new somatic events were found to be acquired at relapse, with the presence of disease-related mutations at 30 days from HSCT associated with a high risk of progression at a median of 67 days from mutation detection.36
In this line, Heuser et al identified mutations of NRAS, U2AF1, IDH2, and TP53 and/or the presence of a complex karyotype as adverse markers of survival in MDS patients undergoing HSCT.37 Although different studies found diverse patterns of gene mutations influencing outcomes, TP53 mutations remain among the major determinants of poor prognosis post HSCT, being consistently associated across studies with reduced survival and a high risk of relapse. In a large cohort of 1514 MDS patients undergoing allogeneic HSCT, patients older than 40 years harboring mutations in TP53 and RAS pathway genes had significantly shorter survival and a higher risk of relapse than their wild-type counterparts.38 While RAS pathway mutations were unfavorable only for patients receiving reduced-intensity conditioning, TP53 mutations were associated with a dismal prognosis irrespective of the intensity of the conditioning regimen. Likewise, TP53 mutations had an impact on the outcome of HSCT when associated with complex karyotype, while RAS pathway mutations were prognostically unfavorable in the context of MDS/myeloproliferative neoplasms in another study.39
Germline mutations
The situation becomes even hazier when considering genes mutated in myeloid neoplasms with germline predisposition, which have been increasingly identified in the last few years (Figure 3). The precise recognition of such variants is important for a correct patient management (especially for genes with other organ involvement, such as Fanconi anemia cases and others), for genetic counseling, and ultimately for donor selection in patients eligible for HSCT (unrelated vs related donors).40 In these cases the variants must be confirmed on a nonhematopoietic tissue (eg, fibroblasts, hair follicles, or nails), and genetic counseling must be required. Generally, these patients present with younger age, a positive family history, and in some instances with extrahematologic features (such as in Emberger syndrome, or MonoMAC in case of GATA2 defects).41 However, a low penetrance may result in an absent family history, and late age at MDS diagnosis (eg, in DDX41-positive cases) may challenge the recognition.42 As demonstrated by a seminal study, these issues play a significant role in the HSCT context, where donor selection becomes of utmost importance. Indeed, the top mutated genes in younger patients (<40 years) included GATA2, PIGA, and SBDS, which are recurrently mutated in myeloid neoplasms with germline predisposition, while mutations in TET2, DNMT3A, SRSF2, and SF3B1 were significantly less common.38

Time course of the identification of genes involved in myeloid neoplasms with germline predisposition. Time line depicting the discovery of the most important genes involved in the development of myeloid disorders with germline predisposition. Different colors represent the associated peculiar features (eg, platelet disorders or organ dysfunction). For gene groups (eg, Fanconi anemia), the year of discovery of the first gene associated with the disease is indicated.
Time course of the identification of genes involved in myeloid neoplasms with germline predisposition. Time line depicting the discovery of the most important genes involved in the development of myeloid disorders with germline predisposition. Different colors represent the associated peculiar features (eg, platelet disorders or organ dysfunction). For gene groups (eg, Fanconi anemia), the year of discovery of the first gene associated with the disease is indicated.
Implications of molecular data for treatment strategy
Several emerging targeted therapies are currently under evaluation for patients with MDS and recurrent gene mutations (Figure 4). For instance, vitamin C is able to restore the metabolic impairment imprinted by TET2 mutations (NCT03682029, NCT03999723) while promising results have been obtained in vitro with splicing inhibitors (eg, H3B-8800),43 but unfortunately, they have not been confirmed by the phase 1, first-in-human clinical study (NCT02841540).

Exemplificative list of genes recurrently mutated in MDS, with impact on clinical features and on treatment options. Shown are exemplary gene mutations, their prognostic impact, the associated recurrent clinical features, and possible therapeutic interventions.
Exemplificative list of genes recurrently mutated in MDS, with impact on clinical features and on treatment options. Shown are exemplary gene mutations, their prognostic impact, the associated recurrent clinical features, and possible therapeutic interventions.
APR-246 is a small molecule able to reestablish p53 functions by restoring its conformation and has shown encouraging clinical results in vivo in combination with HMA.44 A recent phase 1b/2 study of its combination with azacitidine (AZA) in patients with TP53-mutant MDS or AML with 20% to 30% BM blasts showed a 73% overall response rate (ORR), with 50% of patients achieving CR and 58%, a cytogenetic response.45 The efficacy of this combination was also underlined by the reduction of mutational burden and of p53 expression by immunohistochemistry.45 However, the phase 3 of this trial (NCT03745716) missed the primary end point, as the difference in CR between the 2 arms did not meet the predefined threshold for statistical significance (33.3% vs 22.4% in the experimental vs AZA-monotherapy group, respectively; P = .13).46 These data highlight the timeless need to confirm phase 1 and 2 results with larger phase 3 studies even in patients with molecularly defined diseases, which display wide clinical heterogeneity.
Beyond specific TP53 activators, magrolimab, a first-in-class anti-CD47 antibody, has shown particular activity in TP53-mutated MDS/AML, as revealed by the results of a phase 1b trial in combination with AZA in this setting.47 These 2 drugs act synergistically, inducing “eat me” signals on leukemic stem cells and restoring macrophage-mediated phagocytosis.
The recent results of the IDH1/2 inhibitors ivosidenib and enasidenib in AML have also opened new scenarios for patients with MDS.48-50 In a phase 2 study, enasidenib in combination with AZA showed an ORR of 67% in newly diagnosed IDH2-mutant high-risk-MDS patients, and a 50% ORR was obtained when enasidenib is used as a single agent in HMA-treated patients. The preliminary analysis of another study (NCT02074839) currently evaluating ivosidenib in IDH1-mutant cases demonstrated a 42% ORR in relapsed/refractory MDS.51,52 Furthermore, DiNardo and colleagues recently showed that ivosidenib in combination with AZA induced responses in 78% of newly diagnosed patients with IDH1-mutant AML ineligible for intensive therapy and IDH1 mutation clearance in 71% of those achieving CR.50 Another study combining ivosidenib plus venetoclax with or without AZA in the same patient group (NCT03471260) is currently ongoing, and the preliminary results have demonstrated the tolerability and effectiveness of this combination (ORR, 89%).53
Conclusions
MDS is a puzzling disorder with a biological intricacy reflected by the difficulties and limitations of existing classifications and prognostication systems. The abundance of molecular information opens new scenarios in the clinical setting, indeed inaugurating a new era in MDS and delineating a more precise, objective, and personalized path (Figure 5).
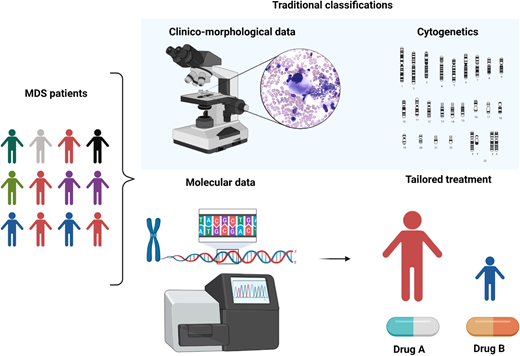
Clinicobiological characterization of MDS and tailored treatment. MDS patients represent a heterogeneous multitude characterized by different clinical, karyotypic, morphologic, and molecular features. In particular, the lower panel demonstrates how the incorporation of molecular information into currently available prognostication schemes will enable in the near future better prognostication and tailored treatments.
Clinicobiological characterization of MDS and tailored treatment. MDS patients represent a heterogeneous multitude characterized by different clinical, karyotypic, morphologic, and molecular features. In particular, the lower panel demonstrates how the incorporation of molecular information into currently available prognostication schemes will enable in the near future better prognostication and tailored treatments.
The bioinformatic interpretation of the clinical role of somatic mutations is still problematic. Nevertheless, machine-learning approaches may in the near future represent a valuable tool to solve the shortcomings of current diagnostic schemes, possibly unveiling further prognostic implications. By combining morphologic and molecular data, these approaches will enable the identification of nonrandom genotypic/morphologic relationships, better defining clinically relevant phenotypes.54 However, a limitation of these studies is demonstrated by the statistical power of the sample size, which does not account for the clinical and prognostic weight of less frequently mutated genes. Larger cohorts of patients will help unravel the complexity of MDS biology with the intent of generating molecularly oriented classification and prognostication systems with relevant clinical utility.
Acknowledgments
We thank the American Italian Cancer Foundation Fellowship Program (to C. G.), AIRC 5 × 1000 call “Metastatic disease: the key unmet need in oncology to MYNERVA” project, #21267 (MYeloid NEoplasms Research Venture AIRC), and MIUR grant N. 2017WXR7ZT (to M. T. V.).
We would also like to acknowledge Stefan Hohaus for helpful comments on the manuscript.
Conflict-of-interest disclosure
Maria Teresa Voso: no competing financial interests to declare.
Carmelo Gurnari: no competing financial interests to declare.
Off-label drug use
Maria Teresa Voso: nothing to disclose.
Carmelo Gurnari: nothing to disclose.
