

Abstract
Myelofibrosis is one of the classical Philadelphia chromosome–negative myeloproliferative neoplasms characterized by progressive marrow failure and chronic inflammation. Discovery of the JAK2 mutation paved the way for development of small molecular inhibitors and further facilitated the research in understanding of molecular biology of the disease. Development of novel medications and synergistic combinations with standard JAK inhibitor (JAKi) therapy may have the potential to improve depth and duration of disease control and symptomatic benefit, whereas advancements in allogeneic hematopoietic stem cell transplantation (HCT) have improved tolerability and donor availability, allowing for more patients to pursue this potentially curative therapy. The increase in options for medical therapy and changing risk profile of HCT is leading to increased complexity in counseling patients on choice of management strategy. In this case-based review, we summarize our approach to symptom-directed medical therapy, including the use of novel drugs and combination therapies currently under study in advanced clinical trials. We outline our recommendations for optimal timing of HCT, including risk-adapted selection for early HCT as opposed to delayed HCT after upfront JAKi therapy, as well as the use of pretransplant JAKi and alternative donor sources.
Learning Objectives
Outline a risk-adapted approach for selection of upfront HCT vs nontransplant therapies for MF in chronic phase
Overview the progress of symptom-directed JAKi therapy toward disease-modifying novel therapies for MF
Introduction
Myelofibrosis (MF) encompasses a group of clonal stem cell disorders that are either primary myelofibrosis or arise from a preceding polycythemia vera (post-PV MF) or essential thrombocythemia (ET; post-ET MF). Although somatic mutations and disease biology may differ between primary and secondary MF, the management approach remains similar. Somatic mutations in myeloproliferative neoplasm (MPN) driver (JAK2, CALR, MPL) and other myeloid malignancy genes perpetuate increased cytokine expression, inflammation, and fibrosis within the niche environment of the bone marrow (BM).1 The clinical manifestations of MF include cytokine-mediated constitutional symptoms (night sweats, weight loss, fever), cytopenias from progressive marrow failure, extramedullary hematopoiesis, and mechanical symptoms from splenomegaly. MF is a heterogenous disease with variable rates of progression to death and blastic transformation depending on underlying clinical and genetic risk factors.
Allogeneic hematopoietic stem cell transplantation (HCT) is the only therapy with potential for cure at present but is limited by significant mortality and morbidity. Accurate estimation of risk to inform patient-centered decision making is a priority concern in managing MF. The approval of JAK inhibitor (JAKi) therapy with ruxolitinib and, more recently, fedratinib provides an alternative effective approach to manage constitutional and splenomegaly-related symptoms in patients with MF who are not candidates or do not wish to pursue HCT. JAKi therapy is not curative and has limited duration of clinical benefit as half of patients experience treatment failure after 2 to 3 years.2,3 A variety of novel agents are in advanced clinical trials for MF, and these emerging therapies may lead to disease modification or improved depth and duration of symptom control.4 Conversely, progress in supportive care, conditioning, graft-versus-host disease (GVHD) prophylaxis, and increasing safety of alternative donor sources may allow for more patients to benefit from HCT.
In this case-based review, we outline our approach and framework for individualized decision making for patients with MF on selection of HCT vs nontransplant strategies, including emerging therapies through clinical trials.
CLINICAL CASES
Case 1: A 60-year old white woman had symptoms of slowly progressive anemia, abdominal discomfort, and early satiety. Physical examination showed a spleen palpable 12 cm below the costal margin (BCM). Hemoglobin was 95 g/L, white blood cell count was 7.0 × 109/L, platelets were 150 × 109/L, and peripheral blood (PB) blasts were 1.2%. Bone marrow biopsy (BMBx) specimen was consistent with diagnosis of overt primary myelofibrosis with World Health Organization grade 3 fibrosis and blasts less than 5%. Cytogenetics showed normal karyotype. Targeted next-generation sequencing demonstrated type 1 CALR mutation as the sole abnormality (variant allele frequency (VAF) 35%).
Case 2: A 63-year-old man of east Indian origin with a long-standing diagnosis of PV lost the need for phlebotomy. He was asymptomatic, and physical examination showed palpable spleen 7 cm BCM. Complete blood count showed the following: hemoglobin, 130 g/L; white blood cell count, 15.3 × 109/L; platelets, 47 × 109/L; and PB blasts, 2%. BMBx specimen was consistent with post-PV MF with World Health Organization grade 2 fibrosis and blasts less than 5%. Cytogenetics demonstrated a deletion of 11q in 15 of 20 metaphases. The following pathogenic mutations were noted in next-generation sequencing studies: JAK2 V617F (VAF 80%), ASXL1 (VAF 27%), and IDH1 (VAF 36%).
Optimal timing of HCT in MF?
Optimal timing of HCT is not well defined, but current expert opinions recommend HCT to patients with MF who have Dynamic International Prognostic Scoring System (DIPSS) intermediate-2/high-risk disease and intermediate-1 risk with additional risk factors.5,6 Transplant studies have reported better outcomes in patients treated earlier in the disease course.7 However, the same group of patients also has better and more durable response with JAKi therapy.8,9 To our knowledge, there is no prospective study to compare the outcomes of HCT vs nontransplant therapies in MF, and limited retrospective studies were either done in the pre-JAKi era or had a very small proportion of patients treated with JAKi.7,10 Lack of comparative data in this area has led to significant variations in practice on application of HCT in MF.
When discussing with a patient regarding the transplant timing, various time points for transplant can be considered (Figure 1):
Upfront transplantation: early HCT usually within a few months of diagnosis
Delayed transplantation: HCT is delayed as long as one can, transplant offered at early signs of failure of nontransplant therapy
Leukemic progression: HCT at time of transformation to acute phase (AP) (PB or BM blasts 10%-19%) or blast phase (BP) (PB or BM blasts ≥20%)

There is no one-size-fits-all approach in patients with MF; however, we strongly recommend against waiting for disease to progress to AP/BP as outcomes after the HCT are poor and many patients never make it to the transplant stage.11-13
Our goal in the beginning is to understand the natural history of the disease based on best available information on clinical and genetic risk factors. This information helps in understanding the expected survival and the likelihood of response and the durability of nontransplant therapy. Patients with an expected survival of less than 5 years and/or less likelihood of durable response from nontransplant therapy are defined as having high-risk MF (Table 1). We recommend the option of upfront HCT in these patients as part of shared decision making incorporating patient fitness, values, and preferences, noting that many patients will choose to delay or forgo transplant following counseling.19,20
Identifying high-risk patients with survival less than 5 years in myelofibrosis
| Risk stratification model | Risk group | Median OS, y | Pros | Cons |
|---|---|---|---|---|
| DIPSS14 | Intermediate-2 | 4.0 | Easy applicability based on clinical/laboratory variables | Does not include any information on genetic variables that may have significant impact on the natural history of the disease |
| High | 1.5 | |||
| DIPSS +15 | Intermediate-2 | 2.9 | Includes cytopenias and cytogenetic data | No mutational data |
| High | 1.3 | |||
| MIPSS70*16 | High | 3.1 | Includes impact of MPN driver genes and HMR† genes | No cytogenetic data |
| MIPSS70 + 2.0*17 | High | 4.1 | Cytogenetic data Driver/HMR‡ genes | |
| Very high | 1.8 | |||
| MPN personalized risk§18 | TP53/–17p/–5/–5q | 2.4 | Combination of clinical/genetic/cytogenetic data | Study heavily weighted toward PV/ET patients Copy number variation not included in most clinical NGS reports |
| Risk stratification model | Risk group | Median OS, y | Pros | Cons |
|---|---|---|---|---|
| DIPSS14 | Intermediate-2 | 4.0 | Easy applicability based on clinical/laboratory variables | Does not include any information on genetic variables that may have significant impact on the natural history of the disease |
| High | 1.5 | |||
| DIPSS +15 | Intermediate-2 | 2.9 | Includes cytopenias and cytogenetic data | No mutational data |
| High | 1.3 | |||
| MIPSS70*16 | High | 3.1 | Includes impact of MPN driver genes and HMR† genes | No cytogenetic data |
| MIPSS70 + 2.0*17 | High | 4.1 | Cytogenetic data Driver/HMR‡ genes | |
| Very high | 1.8 | |||
| MPN personalized risk§18 | TP53/–17p/–5/–5q | 2.4 | Combination of clinical/genetic/cytogenetic data | Study heavily weighted toward PV/ET patients Copy number variation not included in most clinical NGS reports |
HMR (ASXL1, IDH1/2, EZH2, SRSF2).
Includes ASXL1, IDH1/2, EZH2, SRSF2; adds U2AF1 Q157.
NGS, next-generation sequencing.
Evolution of risk assessment in MF
Significant progress has been made in the past decade on understanding the natural history of MF (Figure 2). Early prognostic models incorporated only basic laboratory variables.21,22 Using clinical and laboratory parameters, the International Working Group for Myelofibrosis Research and Treatment developed the International Prognostic Scoring System valid at the time of diagnosis of MF and further validated in the DIPSS applicable at any time during the disease course.14,23 Mayo Clinic investigators refined this score by adding transfusion requiring anemia, thrombocytopenia, and cytogenetics leading to the DIPSS plus score.15 The impact of MPN driver mutations (JAK2, CALR, and MPL) subsequently showed a more indolent course associated with the type 1 CALR mutation, whereas other myeloid malignancy high molecular risk (HMR) mutations, including ASXL1, EZH2, IDH1/2, and SRSF2, were linked with poorer prognosis.26-28 Mutational data alone have been used as the basis for the Genetically Inspired Prognostic Scoring System, whereas integration of all the above factors resulted in the Mutation-Enhanced International Prognostic Scoring System (MIPSS70), which was further refined to include cytogenetic risk in the MIPSS70 plus model.16,24 Mutations in the U2AF1 Q157 hotspot were included as an additional HMR in the MIPSS70 plus v2.0 (http://www.mipss70score.it).17
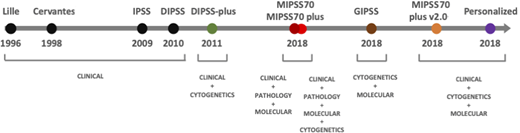
Using a large cohort of 2035 patients with MPN (1321 with ET, 356 with PV, and 309 with primary or secondary MF and 49 with other MPNs), Grinfeld et al18 identified 8 genomic subgroups with distinct clinical and prognostic features. This was used to develop a personalized MPN risk calculator that incorporates clinical, cytogenetic, and mutation data to predict survival and leukemic transformation (https://cancer.sanger.ac.uk/mpn-multistage). A key observation in this study was the poor survival in patients with mutated TP53, a rare yet influential mutation in chronic phase MF but one whose prognostic significance had not been previously described.
Mutation analysis in myeloid neoplasms is a rapidly progressing field, and interactions of various subclonal populations and their downstream effects on clinical outcomes are just beginning to be understood. The definition of HMR mutations will likely evolve further with inclusion of other candidate gene mutations, such as the RAS pathway (NRAS, KRAS, and CBL) and NFE2, which have been shown to confer poorer prognosis in MF.29,30
Survival following HCT has been shown to correlate with DIPSS category, but this does not include transplant-specific variables that may influence clinical outcomes.7 The clinical-molecular MF transplant scoring system, incorporating clinical data, donor type, and mutation status for ASXL1/CALR/MPL, predicts overall survival (OS) and nonrelapse mortality (NRM) in primary and secondary MF.31
We strongly recommend that a physician should use the maximal available information on clinical, laboratory, and genetic parameters depending on the access to the tests. If mutational testing is available, we recommend using the MIPSS70 or MIPSS70 plus v2.0 and otherwise use the DIPSS.
CLINICAL CASES (continued)
In case 1, although the patient had a DIPSS score 3 (ie, intermediate- 2 risk), the tempo of the disease was indolent likely due to the type 1 CALR mutation without additional somatic mutations or cytogenetic abnormalities reflected in a low MIPSS70 + v2.0 risk. Although symptomatic, the patient was at relatively lower risk of significant disease progression, and symptoms were amenable to JAKi therapy; therefore, we would delay the HCT for such a patient until time of JAKi failure. The patient in case 2 had high-risk genetic abnormalities, including an adverse cytogenetic abnormality and 2 HMR mutations. Although the patient had no symptoms and was clinically well, there was high risk of disease progression or leukemic transformation. Therefore, we would refer such a patient for consideration of upfront HCT.
Our algorithmic approach to use of HCT in transplant-eligible patients is summarized in Figure 3.
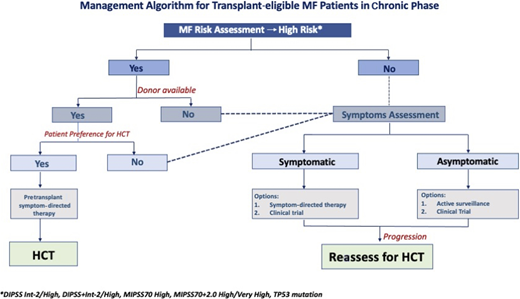
Management algorithm for transplant-eligible patients with MF in the chronic phase.
Management algorithm for transplant-eligible patients with MF in the chronic phase.
Nontransplant therapies in MF
Splenomegaly and constitutional symptoms
Although recommendation for HCT is based on risk assessment, current medical therapy is directed at management of symptoms. Ruxolitinib has demonstrated improvements in splenomegaly, MF-related symptom burden, and health-related quality of life.32,33 Initial trials did not include patients with lower IPSS risk scores, and subsequent studies have shown symptom benefit from ruxolitinib in a lower-risk population.8 Rates of spleen response correlate with ruxolitinib dosing, and cytopenias are the main dose-limiting toxicity. Other JAKi have been studied in phase 3 trials with fedratinib approved by the US Food and Drug Administration for use in MF.34 Momelotinib and pacritinib are other JAKi going through additional phase 3 trials (Table 2) and may have differentiating value for patients with anemia and severe thrombocytopenia, respectively. Factors that predict a shorter duration of response to front-line JAKi therapy include higher DIPSS score, transfusion dependence, number of mutations, and ASXL1/EZH2 mutations specifically.9,35
Ongoing phase 3 trials in myelofibrosis
| Agent | Class of investigational agent | Trial | n | Phase 2 clinical benefit | Primary outcome | Key Secondary outcomes | Comparator | Population/key inclusion criteria |
|---|---|---|---|---|---|---|---|---|
| JAKi-naive patients | ||||||||
| Pelabresib + ruxolitinib | BET inhibitor | MANIFEST-2 | 310 | Spleen reduction Less anemia BM fibrosis reduction | SVR ≥35% at 24 weeks | TSS at 24 weeks | Placebo + ruxolitinib | |
| Navitoclax + ruxolitinib | BCL2 inhibitor | TRANSFORM-1 | 230 | Spleen reduction | SVR ≥35% at 24 weeks | TSS at 24 weeks OS Reduction in BM fibrosis | Placebo + ruxolitinib | |
| Parsaclisib + ruxolitinib | PI3Kδ inhibitor | LIMBER-313 | 440 | Spleen reduction | SVR ≥35% at 24 weeks | TSS OS | Placebo + ruxolitinib | |
| Luspatercept + JAKi | ActRII ligand trap | INDEPENDENCE | 309 | Transfusion independence/anemia improvement | RBC transfusion independence at 24 weeks | Anemia improvement Duration of benefit | Placebo + JAKi | Transfusion dependent |
| Pacritinib | JAKi | PACIFICA | 348 | Spleen reduction Better tolerability in thrombocytopenic patients | SVR ≥35% at 24 weeks | TSS OS | Randomized 2:1 Physician selected BAT | Platelets <50 000/µL Includes patients with prior JAKi exposure |
| Jaktinib | JAKi | — | 105 | SVR ≥35% at 24 weeks | Transfusion dependence | Hydroxyurea | ||
| Following first-line JAKi exposure or failure | ||||||||
| Momelotinib | JAKi | MOMENTUM | 180 | Anemia improvement | TSS at 24 weeks | Transfusion independence Spleen response rate | Randomized 2:1 Danazol | Patients with anemia <100 g/L |
| Fedratinib | JAKi | FREEDOM-2 | 192 | Spleen reduction | SVR ≥35% at 24 weeks | TSS OS | Randomized 2:1 BAT | |
| Navitoclax + ruxolitinib | BCL2 inhibitor | TRANSFORM-2 | 330 | Spleen reduction | SVR ≥35% at 24 weeks | TSS at 24 weeks OS Reduction in BM fibrosis | BAT | |
| Parsaclisib + ruxolitinib | PI3Kδ inhibitor | LIMBER-304 | 212 | Spleen reduction | SVR ≥35% 24 weeks | TSS OS | Placebo + ruxolitinib | |
| Imetelstat | Telomerase inhibitor | IMpactMF | 320 | Symptom score reduction OS | OS | TSS at 24 weeks PFS SVR ≥35% at 24 weeks Reduction in BM fibrosis | BAT | |
| Agent | Class of investigational agent | Trial | n | Phase 2 clinical benefit | Primary outcome | Key Secondary outcomes | Comparator | Population/key inclusion criteria |
|---|---|---|---|---|---|---|---|---|
| JAKi-naive patients | ||||||||
| Pelabresib + ruxolitinib | BET inhibitor | MANIFEST-2 | 310 | Spleen reduction Less anemia BM fibrosis reduction | SVR ≥35% at 24 weeks | TSS at 24 weeks | Placebo + ruxolitinib | |
| Navitoclax + ruxolitinib | BCL2 inhibitor | TRANSFORM-1 | 230 | Spleen reduction | SVR ≥35% at 24 weeks | TSS at 24 weeks OS Reduction in BM fibrosis | Placebo + ruxolitinib | |
| Parsaclisib + ruxolitinib | PI3Kδ inhibitor | LIMBER-313 | 440 | Spleen reduction | SVR ≥35% at 24 weeks | TSS OS | Placebo + ruxolitinib | |
| Luspatercept + JAKi | ActRII ligand trap | INDEPENDENCE | 309 | Transfusion independence/anemia improvement | RBC transfusion independence at 24 weeks | Anemia improvement Duration of benefit | Placebo + JAKi | Transfusion dependent |
| Pacritinib | JAKi | PACIFICA | 348 | Spleen reduction Better tolerability in thrombocytopenic patients | SVR ≥35% at 24 weeks | TSS OS | Randomized 2:1 Physician selected BAT | Platelets <50 000/µL Includes patients with prior JAKi exposure |
| Jaktinib | JAKi | — | 105 | SVR ≥35% at 24 weeks | Transfusion dependence | Hydroxyurea | ||
| Following first-line JAKi exposure or failure | ||||||||
| Momelotinib | JAKi | MOMENTUM | 180 | Anemia improvement | TSS at 24 weeks | Transfusion independence Spleen response rate | Randomized 2:1 Danazol | Patients with anemia <100 g/L |
| Fedratinib | JAKi | FREEDOM-2 | 192 | Spleen reduction | SVR ≥35% at 24 weeks | TSS OS | Randomized 2:1 BAT | |
| Navitoclax + ruxolitinib | BCL2 inhibitor | TRANSFORM-2 | 330 | Spleen reduction | SVR ≥35% at 24 weeks | TSS at 24 weeks OS Reduction in BM fibrosis | BAT | |
| Parsaclisib + ruxolitinib | PI3Kδ inhibitor | LIMBER-304 | 212 | Spleen reduction | SVR ≥35% 24 weeks | TSS OS | Placebo + ruxolitinib | |
| Imetelstat | Telomerase inhibitor | IMpactMF | 320 | Symptom score reduction OS | OS | TSS at 24 weeks PFS SVR ≥35% at 24 weeks Reduction in BM fibrosis | BAT | |
BAT, best available therapy; BCL2, B-cell lymphoma 2; BET, bromodomain and extraterminal; PFS, progression-free survival; SVR, spleen volume response; TSS, total symptom score.
Source: https://www.clinicaltrials.gov accessed on April 30, 2021.
Deepening and prolonging the spleen and symptom response is a primary goal of many novel combination strategies adding additive/synergistic agents to ruxolitinib in symptomatic treatment-naive patients (Table 2). In addition to the constitutive activation of JAK-STAT pathways, there is evidence that cytokine burden, disease progression, and JAKi resistance can be affected through aberrant activation of other pathways that mediate cell proliferation, survival, and cytokine signaling.1,4 Promising agents used in combination with ruxolitinib currently in phase 3 evaluation are summarized in Table 2. Pelabresib is a bromodomain and extra-terminal domain inhibitor that reduces nuclear factor–κB–mediated inflammation. Navitoclax is a BCL-2/BCL-XL inhibitor that reduces prosurvival signaling and promotes apoptosis in the malignant clone. Parsaclisib, a phosphatidylinositol 3-kinase (PI3K) δ inhibitor, reduces abnormal PI3K/mTOR/AKT activation that promotes inflammation and JAKi resistance. In phase 2 studies, combinations of these agents with ruxolitinib have exhibited good rates of spleen reduction and symptom score improvement, with pelabresib and navitoclax containing regimens demonstrating evidence of reduced BM fibrosis in some patients.4 Pelabresib, navitoclax, and parsaclisib combinations with ruxolitinib are being compared with ruxolitinib monotherapy in treatment-naive patients in the phase 3 MANIFEST-2, TRANSFORM-1, and LIMBER-313 trials, respectively.
Cytopenias
Management of cytopenias is an unmet need in MF, both in otherwise asymptomatic patients and in those whose JAKi treatment is complicated by anemia and thrombocytopenia. Supportive care relies on transfusion of red blood cells and platelets, whereas pharmacologic therapy is limited to danazol and erythropoiesis-stimulating agents. Single-agent pelabresib is being investigated for improving anemia as monotherapy for patients who do not have an indication for JAKi therapy.4 Enhancement of late-stage erythrocyte development through the inhibition of transforming growth factor β/SMAD signaling with activin receptor ligand traps, such as luspatercept, has demonstrated efficacy in improving anemia and reducing transfusion, and it will be evaluated further in the phase 3 INDEPENDENCE study (Table 2).
Asymptomatic patients
There is currently no medical therapy that has clear benefit in asymptomatic patients; we only consider it in the setting of a clinical trial for an agent with disease-modifying potential. Immune therapies, including interferon agents, and antifibrotic agents such as PRM-151 have shown some promise in early studies but require confirmation in controlled trials.4,36-38
CLINICAL CASES (continued)
In case 1, the patient responded well to upfront ruxolitinib with resolution of symptoms and reduction in her spleen size to a nadir of 6 cm BCM. Such a patient could also be considered for an upfront combination therapy trial.
After 6 years, her spleen began to increase in size again, up to 15 cm BCM, with recurrence of abdominal pain and early satiety. The patient was started on fedratinib and had significant improvement in spleen size. Subsequently, this patient underwent 10/10 matched unrelated donor (MUD) transplant using reduced-intensity conditioning and remains well after HCT.
JAKi failure
Prior studies evaluating outcomes following discontinuation of ruxolitinib report a median survival of 11 to 16 months (Table 3), with very short survival in those with AP/BP transformation.39-42 Although informative, we note that in real-world practice, many patients continue on ruxolitinib despite clear evidence of disease progression due to the lack of alternative medical therapy. With the approval of fedratinib and availability of novel combination strategies as part of clinical trials (Table 2), early recognition of JAKi therapy failure and its pattern is of paramount importance for selecting appropriate options for these patients. We use the criteria proposed by the Canadian MPN Group and classify the JAKi failure by categories such as loss of spleen/symptom response, severe cytopenias, or progression to AP/BP.43
Survival and predictive factors following ruxolitinib failure/discontinuation
| Reference | Median OS (95% CI) following ruxolitinib failure, mo | Factors predicting lower survival |
|---|---|---|
| Newberry et al. (2017)39 | 14 (10-18) | Platelets <260 at ruxolitinib start Platelets <100 at discontinuation Emergent mutations at discontinuation |
| Kuykendall et al. (2018)40 | 13 | Lack of salvage therapy |
| Mascarenhas et al. (2020)41 | 11.1 (8.4-14.5) | Age >65 years Charlson Comorbidity Index |
| Palandri et al. (2020)42 | 13.2 (8.0-22.7) | Blast phase at failure Hb <100 g/L Peripheral blasts >1% |
| Reference | Median OS (95% CI) following ruxolitinib failure, mo | Factors predicting lower survival |
|---|---|---|
| Newberry et al. (2017)39 | 14 (10-18) | Platelets <260 at ruxolitinib start Platelets <100 at discontinuation Emergent mutations at discontinuation |
| Kuykendall et al. (2018)40 | 13 | Lack of salvage therapy |
| Mascarenhas et al. (2020)41 | 11.1 (8.4-14.5) | Age >65 years Charlson Comorbidity Index |
| Palandri et al. (2020)42 | 13.2 (8.0-22.7) | Blast phase at failure Hb <100 g/L Peripheral blasts >1% |
Pretransplant JAKi therapy
In patients referred for HCT, massive splenomegaly has been associated with delayed count recovery and possibly worse OS.44 Limited evidence is available to define an appropriate target spleen size, although splenectomy is considered in patients with massive splenomegaly that is refractory to medical therapy.44,45
Use of peritransplant JAKi has significant interest given the potential for both disease control and impact on GVHD (Table 4). Preliminary results from the JAK-ALLO trial raised safety concerns with tumor lysis syndrome and cardiogenic shock, potentially due to cytokine flare following sudden cessation of ruxolitinib, necessitating a pause on the study.46 However, subsequent trials do not suggest substantial risk to this approach.47,48 The JAK-ALLO results demonstrated improved safety, with no tumor lysis syndrome or ruxolitinib withdrawal syndrome, following protocol amendment to an abrupt ruxolitinib discontinuation prior to conditioning.46 The impact of pretransplant JAKi on posttransplant outcomes, including GHVD, is not clear, with retrospective studies demonstrating similar outcomes in those with or without prior JAKi therapy.49 We recommend using JAKi therapy prior to HCT in patients with significant MF-related symptoms (Figure 3), with a tapering schedule leading up to, or a few days after, the start of conditioning to avoid any withdrawal symptoms.46,47,50
Prospective studies of pre-HCT ruxolitinib therapy
| Reference | Number of patients recruited | Number of patients who underwent HCT | Splenectomy prior to HCT, n (%) | Taper | Ruxolitinib discontinuation | SAE during ruxolitinib taper | Conditioning | OS at 2 y, % | NRM at 2 y, % | GF, % | GVHD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gupta et al. (2019)47 | 21 | 19* 5 MSD 14 UD | 0 (0) | 4 days | 1 day prior to start of conditioning | None | RIC (Flu-Bu) | 63 | 28% | 16 | aGVHD grade 3-4, 16% cGVHD 76% at 2 y |
| Salit et al. (2020)48 | 34 | 28† 14 MRD 11 UD 3 UCB | 3 (11) | 5 mg every 3 days | After 2-3 days of conditioning therapy | None | Multiple | 86 | 7% | 0 | aGVHD grade 3-4, 22% cGVHD, 41% |
| Robin et al. (2021)46 | 64 | 59‡ 18 MSD 32 MUD 14 UD 9/10 | 19 (30) | 15 days (n = 17) | 1 day prior to start of conditioning | TLS 3§ RWS 3 Heart failure | RIC (Flu-Mel) | 55 | 46% 23% MSD 50% MUD 77% UD 9/10 | 0 | aGVHD grade 3-4, 44% cGVHD 37% at 2 y |
| None (n = 42) | 1 day prior to start of conditioning | Heart failure |
| Reference | Number of patients recruited | Number of patients who underwent HCT | Splenectomy prior to HCT, n (%) | Taper | Ruxolitinib discontinuation | SAE during ruxolitinib taper | Conditioning | OS at 2 y, % | NRM at 2 y, % | GF, % | GVHD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gupta et al. (2019)47 | 21 | 19* 5 MSD 14 UD | 0 (0) | 4 days | 1 day prior to start of conditioning | None | RIC (Flu-Bu) | 63 | 28% | 16 | aGVHD grade 3-4, 16% cGVHD 76% at 2 y |
| Salit et al. (2020)48 | 34 | 28† 14 MRD 11 UD 3 UCB | 3 (11) | 5 mg every 3 days | After 2-3 days of conditioning therapy | None | Multiple | 86 | 7% | 0 | aGVHD grade 3-4, 22% cGVHD, 41% |
| Robin et al. (2021)46 | 64 | 59‡ 18 MSD 32 MUD 14 UD 9/10 | 19 (30) | 15 days (n = 17) | 1 day prior to start of conditioning | TLS 3§ RWS 3 Heart failure | RIC (Flu-Mel) | 55 | 46% 23% MSD 50% MUD 77% UD 9/10 | 0 | aGVHD grade 3-4, 44% cGVHD 37% at 2 y |
| None (n = 42) | 1 day prior to start of conditioning | Heart failure |
One patient with disease progression to AML, 1 patient with sudden death during ruxolitinib phase.
Three patients with progression to AML during ruxolitinib phase, 3 patients not meeting criteria for transplant.
One donor withdrawal. One patient continued taking ruxolitinib, 3 patients died without HCT; 2 with heart failure following splenectomy.
No TLS following protocol amendment to abrupt ruxolitinib discontinuation.
aGVHD, acute GVHD; AML, acute meloid leukemia; cGVHD, chronic GVHD; Flu-Bu, fludarabine/busulfan; Flu-Mel, fludarabine-melphalan; GF, graft failure; RIC, reduced-intensity conditioning; RWS, ruxolitinib withdrawal syndrome; SAE, significant adverse event; TLS, tumor lysis syndrome; UCB, umbilical cord blood.
CLINICAL CASES (continued)
In case 2, the patient was referred for transplant, but no fully matched sibling donors (MSDs) or unrelated donors (UDs) could be identified. The patient preferred not to pursue haploidentical transplant initially. However, there was increase in blast counts in PB to 8% within 6 months, and the patient opted for haploidentical HCT with his son as a donor at that time point.
Role of alternative donor transplant in MF
HCT from a matched sibling or unrelated donor is the preferred source for HCT in MF, with many early studies demonstrating inferior outcomes using a mismatched unrelated donor.51,52 Recent reporting trends from the Center for International Blood and Marrow Transplant Research show slowly increasing adaption in the use of haploidentical donors in MF as advancements in supportive care and the prevention of GVHD, including the use of posttransplant cyclophosphamide, have allowed for improved safety (Table 5).25 A major limitation of all these studies is small sample size. Bregante et al53 report the increasing use of haploidentical donors from 2011 to 2014 compared with 2000 to 2010, with similar outcomes compared with MSDs among the alternative donor group (77% haploidentical) in the later time period. Initial results from Kunte et al54 also show encouraging outcomes in a retrospective cohort of haploidentical HCT with posttransplant cyclophosphamide. Data following the use of related donors with 2 or more antigen mismatches have been reported from the European Society for Blood and Marrow Transplantation registry demonstrating reasonable 2-year OS (56%) and GVHD rates but higher rates of graft failure (22%) and NRM at 2 years (38%).55 Use of unrelated cord blood also presents similar concerns, with a report from the Japan Society for Hematopoietic Cell Transplantation demonstrating lower rates of engraftment, high NRM, and only 27% 4-year OS.56 Limited data on haploidentical transplants support the potential use in selected patients, although many patients may opt for medical therapy when counseled on the added risks associated with alternative donors.15 Ongoing large studies through the Center for International Blood and Marrow Transplant Research comparing outcomes of haploidentical transplants vs MUDs and mismatched donors should further clarify and support the positioning of haploidentical transplants in the management of MF (Tania Jain, Johns Hopkins University, personal communication, 2021).
HCT using alternative donors in myelofibrosis
| Reference | Donor type | n | Conditioning | GVHD prophylaxis | OS (95% CI) | NRM (95% CI) | Graft failure | GVHD |
|---|---|---|---|---|---|---|---|---|
| Raj et al. (2019)55 | Mismatched related donor | 56 | MAC (70%)/RIC (30%) | PTCy in 79% | 56% (41%-70%) at 2 y | 38% (24%-51%) at 2 y | Primary 9% Secondary 13% | aGVHD* 28% cGVHD 45% at 1 y |
| Murata et al. (2019)56 | Unrelated cord blood | 13 | — | — | 48% at 1 y 27% at 4 y | 41% (22%-60%) at 1 y 62% (35%-81%) at 4 y | — | aGVHD* 31% cGVHD 15% at 1 y |
| Kunte et al. (2020)54 | Haploidentical | 58 | NMA (52%)/RIC (45%) | PTCy in 100% | 69% (55%-80%) at 2 y | 21% (11%-34%) at 2 y | Primary 9% | aGVHD* 44% at 6 m cGVHD 31% at 2 y |
| Reference | Donor type | n | Conditioning | GVHD prophylaxis | OS (95% CI) | NRM (95% CI) | Graft failure | GVHD |
|---|---|---|---|---|---|---|---|---|
| Raj et al. (2019)55 | Mismatched related donor | 56 | MAC (70%)/RIC (30%) | PTCy in 79% | 56% (41%-70%) at 2 y | 38% (24%-51%) at 2 y | Primary 9% Secondary 13% | aGVHD* 28% cGVHD 45% at 1 y |
| Murata et al. (2019)56 | Unrelated cord blood | 13 | — | — | 48% at 1 y 27% at 4 y | 41% (22%-60%) at 1 y 62% (35%-81%) at 4 y | — | aGVHD* 31% cGVHD 15% at 1 y |
| Kunte et al. (2020)54 | Haploidentical | 58 | NMA (52%)/RIC (45%) | PTCy in 100% | 69% (55%-80%) at 2 y | 21% (11%-34%) at 2 y | Primary 9% | aGVHD* 44% at 6 m cGVHD 31% at 2 y |
Acute GVHD encompasses grades II to IV.
MAC, myeloablative conditioning; NMA, non-myeloablative; PTCy, posttransplant cyclophosphamide.
Conclusion
Advancements in both medical therapy and HCT optimization have improved and broadened therapeutic options for patients with MF. We recommend a risk-adapted strategy for timing of HCT and symptom-directed approach to medical therapy. These recommendations should be incorporated as part of shared decision making encompassing patient preferences and values to decide on an individualized approach in each patient. Where possible, efforts should be made to enroll these patients in prospective trials or registries for better understanding of comparative outcomes.
Acknowledgments
This project was supported by a grant from the Elizabeth and Tony Comper Foundation and the Princess Margaret Cancer Centre Foundation (V.G.).
Conflict-of-interest disclosure
James England: No competing financial interests to declare.
Vikas Gupta: Consultancy and advisory board: Novartis, BMS- Celgene, Sierra Oncology, Pfizer, Abb Vie, Roche.
Off-label drug use
James England: Off-label use of ruxolitinib in the peri-transplant period is discussed in the article.
Vikas Gupta: Off-label use of ruxolitinib in the peri-transplant period is discussed in the article.
