

Abstract
A common feature of both congenital and acquired forms of bone marrow failure is an increased risk of developing acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Indeed, the development of MDS or AML is now the major cause of mortality in patients with congenital neutropenia. Thus, there is a pressing clinical need to develop better strategies to prevent, diagnose early, and treat MDS/AML in patients with congenital neutropenia and other bone marrow failure syndromes. Here, we discuss recent data characterizing clonal hematopoiesis and progression to myeloid malignancy in congenital neutropenia, focusing on severe congenital neutropenia (SCN) and Shwachman-Diamond syndrome. We summarize recent studies showing excellent outcomes after allogenic hematopoietic stem cell transplantation for many (but not all) patients with congenital neutropenia, including patients with SCN with active myeloid malignancy who underwent transplantation. Finally, we discuss how these new data inform the current clinical management of patients with congenital neutropenia.
Learning Objectives
Understand the indications for and interpretation of somatic gene pane sequencing and karyotyping in congenital neutropenia
Integrate recent improvements in hematopoietic stem cell transplantation outcome in congenital neutropenia into clinical management
Appreciate the contribution of stressors in the development of clonal hematopoiesis and myeloid malignancy in congenital neutropenia
Introduction
There has been a recent increase in the use of somatic gene panel sequencing in patients with congenital neutropenia to identify patients at risk of leukemic transformation. We present three cases of congenital neutropenia in which results of molecular testing raise important clinical management questions. To answer these questions, we review recent advances on the following topics: (1) the frequency and molecular features of myeloid malignancy in congenital neutropenia, (2) clonal hematopoiesis in congenital neutropenia and mechanisms of clonal evolution to myeloid malignancy, and (3) recent studies of hematopoietic stem cell transplantation (HSCT) outcomes in congenital neutropenia. Finally, we explore how these new data inform the clinical management of congenital neutropenia, including the case presentations.
CLINICAL CASE 1
This patient presented in early childhood with recurrent infections and an absolute neutrophil count (ANC) of <200 cells/mm3. Bone marrow biopsy specimen showed myeloid maturation arrest, and genetic analysis revealed a heterozygous germline pathogenic variant in ELANE (p.S126L). Serial surveillance bone marrow biopsy specimens demonstrated no overt clonal abnormalities as detected by karyotype and fluorescence in situ hybridization. However, somatic sequencing of a surveillance bone marrow sample obtained at age 45 years showed a high variant allele frequency (41%) truncating pathogenic variant in CSF3R. Lower frequency clones in DNMT3A (1.7%) and ASXL1 (2.6%) also were detected.
CLINICAL CASE 2
This patient presented at age 2 years with neutropenia, failure to thrive with chronic diarrhea, mild developmental delay, and pelvic dysostosis. The ANC was 320 cells/mm3, hemoglobin was 11.7 g/dL, and platelets were 132 000 cells/mm3. Initial bone marrow biopsy performed at age 2 years demonstrated hypocellularity with a decrease in mature myeloid cells and normal cytogenetics. Sequencing revealed biallelic germline pathogenic variants in SBDS (c. 183_184TA _CT and c. 258 + 2T>C), confirming the diagnosis of Shwachman-Diamond syndrome (SDS). Over time, the patient developed moderate anemia with macrocytosis and continued with intermittent mild to moderate neutropenia. Repeat bone marrow examination revealed mild dysmorphology predominantly in the myeloid lineages without dysplasia and megakaryocyte hypoplasia. Serial somatic gene panel sequencing over a 5-year time span identified multiple heterozygous low-abundance TP53 pathogenic missense variants.
CLINICAL CASE 3
The patient presented in infancy with omphalitis and was noted to have severe neutropenia with an ANC of <200 cells/mm3. Before initiation of granulocyte colony-stimulating factor (G-CSF), the patient experienced recurrent sinopulmonary infections and cellulitis. Germline sequencing revealed a pathogenic ELANE variant (p.V65F). Somatic gene panel sequencing obtained at age 7 years did not demonstrate any severe congenital neutropenia (SCN)–associated or other clonal hematopoiesis variants, and serial bone marrow examinations did not reveal any dysplasia or chromosomal abnormalities. Although infectious complications initially improved while on G-CSF therapy, the patient eventually experienced severe neutropenia again with concomitant recurrent cellulitis despite escalating doses of G-CSF (>10 µg/kg/dose).
Myeloid malignancy in congenital neutropenia
SCN is a rare syndrome characterized by chronic neutropenia present from birth and recurring bacterial infections.1 Pathogenic variants of ELANE are the most common cause of SCN, accounting for approximately 50% of cases, with variants in CLPB, HAX1, and G6PC3 accounting for an additional 10% to 20% of cases depending on geographic location (eg, HAX1 variants have not been observed in North America).2-5 Treatment with G-CSF is the standard of care for SCN; it increases the level of circulating neutrophils in most patients and reduces infection-related mortality.6 The development of myeloid malignancy is now the major cause of mortality in SCN. Indeed, after 15 years of G-CSF therapy, the cumulative incidence of myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) in patients with SCN was estimated at 22%.7 In this study, transformation to MDS/AML was associated with a higher G-CSF dose (>8 µg/kg/d) and with a reduced neutrophil response to G-CSF. Similar data from the French Neutropenia Registry reported an 11% cumulative incidence of MDS/AML by age 20 years.8 Of note, almost all genetic subtypes of SCN have been associated with an increased risk for transformation to MDS/AML.9 MDS/AML arising in the setting of SCN is associated with distinct molecular features compared with de novo MDS/AML.10 Most notably, somatic truncating variants in CSF3R and missense variants in RUNX1 are observed in ~80% and 63% of cases, respectively.11 In addition, secondary MDS/AML in patients with SCN is frequently associated with monosomy 7 and trisomy 21.12-14
SDS is an inherited bone marrow failure syndrome characterized by a constellation of extra-hematopoietic manifestations with high variability (exocrine pancreatic insufficiency, hepatopathy, bony abnormalities, poor growth, and other endocrine manifestations).15,16 Neutropenia is present in approximately 90% of cases of SDS, with anemia, thrombocytopenia, and pancytopenia observed less frequently. SDS is caused, in most cases, by biallelic germline variants in SBDS.17 The estimates of risk for hematopoietic malignancy vary from 12% to 20% depending in part on the age of the cohort.8,18,19 MDS/AML that arises in the setting of SDS is characterized by biallelic TP53 variants and complex chromosomal abnormalities.8,20,21
Clonal hematopoiesis in congenital neutropenia
During hematopoiesis, the acquisition of somatic variants over time leads to the development of a genetically heterogeneous blood cell population. Some of these variants confer a fitness advantage, leading to the expansion of hematopoietic stem/progenitor cells (HSPCs) carrying certain variants over time in a process known as clonal hematopoiesis.22,23 Variants in genes implicated in myeloid malignancy are the most frequent in age-related clonal hematopoiesis (ARCH), including in the epigenetic modifiers DNTM3A, TET2, and ASXL1. Of note, in otherwise healthy persons, the presence of clonal hematopoiesis confers an increased risk of developing hematopoietic malignancy, depending on the gene involved, clone size, and whether multiple variants are present.24
Recent studies demonstrate that the frequency of clonal hematopoiesis in patients with congenital neutropenia is markedly increased. We showed that in both SCN and SDS, the incidence of clonal hematopoiesis was markedly increased compared with age-matched healthy donors.25 However, distinct genes were responsible for this increase. In SCN, the increase in clonal hematopoiesis is entirely due to truncating variants in CSF3R; no increase in clonal hematopoiesis due to other genes commonly mutated in clonal hematopoiesis (eg, DNMT3A, TET2, and ASXL1) was observed (Figure 1). In contrast, in SDS, the increase in clonal hematopoiesis is due to a marked increase in TP53 variants, with nearly 50% of patients carrying 1 or more TP53 variants. Of note, TP53 variants were not seen in patients with SCN or in age-matched healthy donors. Conversely, CSF3R variants were not seen in patients with SDS or age-matched healthy donors.

Graphical representation of the observed clonal hematopoiesis rates by somatic genetic alteration within healthy age-matched controls, patients with SCN, or patients with SDS.
Graphical representation of the observed clonal hematopoiesis rates by somatic genetic alteration within healthy age-matched controls, patients with SCN, or patients with SDS.
What accounts for the striking difference in clonal hematopoiesis between persons with SCN and SDS? Current evidence points to differences in cellular stressors in healthy persons and persons with SCN or SDS. The CSF3R variants in patients with SCN typically produce a truncated G-CSF receptor, which transmits a sustained, dysregulated signal in response to G-CSF.26-28 Studies in mice show that expression of a truncated G-CSF receptor confers a competitive advantage to HSPCs that is dependent on chronic G-CSF treatment.29 Thus, the high level of G-CSF present in patients with SCN (either through endogenous production or pharmacologic administration) may drive the expansion of HSPCs carrying truncation CSF3R variants (Figure 2). Interestingly, a recent study examined clonal hematopoiesis in 185 patients with chronic idiopathic neutropenia (also termed idiopathic neutropenia of undetermined significance–neutropenia).30 Clonal hematopoiesis was identified in 21 of 185 (11.4%) patients, which is similar to that expected for age- matched controls. The most frequent somatic variants observed were in DNMT3A and TET2, as observed in ARCH. In sharp contrast to SCN, somatic CSF3R variants were not detected in patients with chronic idiopathic neutropenia. Whether this difference is due to the severity of neutropenia or secondary to cell-intrinsic changes related to germline variants in SCN will require further study.
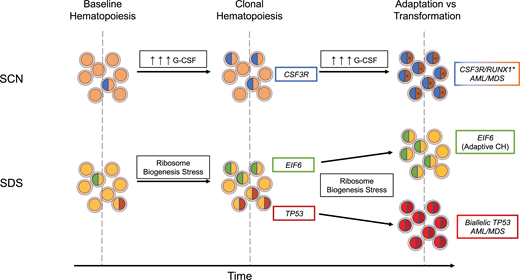
Modes of evolution from clonal hematopoiesis to malignant transformation. In the presence of high exogenous or endogenous G-CSF, subclones with truncating CSF3R variants have a competitive advantage and expand over time. With continued pressure from excessive G-CSF signaling, cooperating RUNX1 variants (*) can contribute to malignant transformation. In SDS, ribosome biogenesis stress selects for subclones that have adapted through acquisition of EIF6 or TP53 somatic variants. Chronic ribosome biogenesis stress can lead to either continued adaptive clonal hematopoiesis or, in TP53 subclones clones that have acquired a second TP53 variant on the other allele, malignant transformation.
Modes of evolution from clonal hematopoiesis to malignant transformation. In the presence of high exogenous or endogenous G-CSF, subclones with truncating CSF3R variants have a competitive advantage and expand over time. With continued pressure from excessive G-CSF signaling, cooperating RUNX1 variants (*) can contribute to malignant transformation. In SDS, ribosome biogenesis stress selects for subclones that have adapted through acquisition of EIF6 or TP53 somatic variants. Chronic ribosome biogenesis stress can lead to either continued adaptive clonal hematopoiesis or, in TP53 subclones clones that have acquired a second TP53 variant on the other allele, malignant transformation.
Germline variants in SBDS that are present in most cases of SDS result in impaired ribosome biogenesis, which in turn induces p53 expression and growth arrest.31-33 Pathogenic somatic variants in TP53 in HSPCs are predicted to attenuate this growth arrest, resulting in their selective expansion in patients with SDS. An elegant study by Kennedy et al20 provides further support for the key role of impaired ribosome biogenesis in the development of clonal hematopoiesis in patients with SDS. Specifically, in addition to TP53 variants, patients with SDS frequently develop inactivating variants of EIF6. These variants partially rescue the ribosome biogenesis defect in SDS, thereby reducing p53 activation. Together, these observations illustrate the importance of cellular stressors in the development of clonal hematopoiesis (and likely myeloid malignancy). In SCN, elevated levels of G-CSF drive the expansion of HSPCs carrying truncation CSF3R variants, and in SDS, impaired ribosome biogenesis selects for HSPCs carrying TP53 variants or variants (ie, EIF6) that reduce p53 activation (Figure 2).
Clonal evolution to myeloid malignancy in congenital neutropenia
The presence of clonal hematopoiesis in patients with congenital neutropenia does not inevitably lead to myeloid malignancy. Although the presence of truncating CSF3R variants is associated with the development of myeloid malignancy in patients with SCN, it is not sufficient. Indeed, longitudinal sequencing studies show that CSF3R-mutant clonal hematopoiesis can persist for many years without progression, even in cases with large CSF3R- mutant clones.34 There is evidence that somatic loss-of-function variants of RUNX1 may be an early progression event, with acquisition of chromosomal abnormalities (monosomy 7 and trisomy 21) late events.11 Consistent with this conclusion, Dannenmann et al35 used SCN patient-derived induced pluripotent stem cells that expressed mutant CSF3R with our without mutant RUNX1 to show that these variants cooperate to block granulocytic differentiation, producing an AML-like phenotype.
Variants in TP53 are present in nearly 50% of patients with SDS and may persist for many years without progression.20,25 Thus, having a TP53 variant within the hematopoietic compartment is clearly not sufficient to drive leukemogenesis. We suggest that continued ribosome biogenesis stress in SDS HSPCs carrying a heterozygous pathogenic TP53 variant selects for clones that inactivate the second TP53 allele (Figure 2). In contrast, variants in EIF6, by reducing ribosome biogenesis stress, may be protective. Consistent with this model, biallelic TP53 variants, but not EIF6 variants, are very common in the malignant clone in patients with SDS who develop myeloid malignancy.20
HSCT in congenital neutropenia
Because the development of myeloid malignancies is the major cause of death in congenital neutropenia, a key question in the clinical management is when to offer HSCT.
Current indications for HSCT in patients with SCN include G-CSF refractoriness or development of MDS/AML, with some investigators also considering high-dose G-CSF requirement (>15 µg/kg/d)36 or certain high-risk ELANE pathogenic variants (p.G214R, p.C151Y, and p.C223ter).37,38 Studies of outcomes in patients with SCN after HSCT are confounded by the different eras of transplant, conditioning regimens, and donor sources. Nonetheless, recent reports39,40 demonstrate that outcomes are good for patients who undergo transplantation with or without myeloid malignancy (Table 1). Also highlighted in these studies is the remarkably low relapse rate after HSCT in patients with SCN who have myeloid malignancy, especially considering that many patients with SCN have active myeloid malignancy at the time of transplant. The reason for the very low relapse rate is uncertain but suggests that SCN leukemic stem cells have reduced fitness and/or increased susceptibility to allogenic immune cell clearance.
Overall survival following HSCT with or without malignancy in modern-era trials
| Post-HSCT overall survival | Without malignancy | With malignancy |
|---|---|---|
| SCN | ||
| *EBMT39 | 87% (n = 73) | 79% (n = 14) |
| *SCNIR (European)40 | 78% (n = 27) | 83% (n = 24) |
| SDS | ||
| ^CIBMTR42 | 72% (n = 39) | 15% (n = 13) |
| ^SAAWP-EBMT41 | 71% (n = 61) | 29% (n = 13) |
| *SDSR43 | — | 46% (n = 15) |
| Post-HSCT overall survival | Without malignancy | With malignancy |
|---|---|---|
| SCN | ||
| *EBMT39 | 87% (n = 73) | 79% (n = 14) |
| *SCNIR (European)40 | 78% (n = 27) | 83% (n = 24) |
| SDS | ||
| ^CIBMTR42 | 72% (n = 39) | 15% (n = 13) |
| ^SAAWP-EBMT41 | 71% (n = 61) | 29% (n = 13) |
| *SDSR43 | — | 46% (n = 15) |
Three-year overall survival; ^five-year overall survival.
CIBMTR, Center for International Blood and Marrow Transplant Research; EBMT, European Society for Bone Marrow Transplantation; SAAWP-EMBT, Severe Aplastic Anemia Working Party of the EBMT; SCNIR, Severe Chronic Neutropenia International Registry; SDSR, Shwachman-Diamond Syndrome Registry of North America.
HSCT for patients with SDS is generally performed for bone marrow failure or myeloid malignancy. HSCT outcomes are inferior to that that seen with patients with SCN and remains especially dismal for those patients who have already developed a myeloid malignancy41-43 (Table 1). The reasons for the inferior outcomes are likely multifactorial, including impaired bone marrow stromal cell function, nonhematopoietic organ toxicity, and, most important, the intrinsic resistance of TP53 mutant myeloid malignancies to therapy. Together, these data suggest that an effective method to identify patients with SDS at high risk of progression to myeloid malignancy for early HSCT is warranted. However, this must be balanced by the likelihood of increased transplant-related morbidity and mortality in patients with SDS who have underlying extra-hematopoietic organ dysfunction.
Timing of HSCT in SCN
There are a number of clinical and molecular data to consider regarding timing of transplant for patients with SCN, including (1) response to G-CSF, (2) results of surveillance bone marrow studies to identify dysplasia or chromosomal abnormalities, and (3) somatic variant detection. Prior studies have demonstrated that G-CSF dose and neutrophil response are predictors of progression to myeloid malignancy.7,19 Based on these data, the French Severe Neutropenia Registry recommended that transplant be offered to all patients with SCN requiring high-dose G-CSF (>15 µg/kg/d). Since instituting this policy in 2005, none of the patients in this cohort have developed a myeloid malignancy.36 Given the excellent HSCT outcomes for patients with SCN who have myeloid malignancy, a wait-and-see approach for such patients also is reasonable. Of note, an inadequate neutrophil response (ANC <1000 cells/mm3) to G-CSF, despite high G-CSF dose, is a strong indication for HSCT.
It is common practice to perform annual bone marrow biopsy evaluations on patients with SCN to identify chromosomal abnormalities and/or dysplasia as early signs of malignant transformation.44-46 Historically, this practice was based on expert opinion rather than strong clinical or experimental evidence. However, bone marrow biopsy is an invasive and expensive procedure, and it also carries associated risks of sedation in younger patients. In our opinion, in light of recent data showing good outcomes for patients with SCN who have myeloid malignancy, the value of annual surveillance bone marrows is debatable. That said, a bone marrow biopsy for patients with SCN with worsening blood counts or decreased responsiveness to G-CSF is warranted.
There has been a recent increased use of gene panel sequencing to identify somatic variants in patients with SCN. An advantage of this approach is the ability to analyze blood samples, obviating the needed for an invasive bone marrow biopsy. On the other hand, the test is expensive and insurance coverage variable. Interpreting the sequencing results also is challenging. Truncating CSF3R variants in SCN are common and may persist for years or even disappear with leukemic progression.34 Thus, the value of truncating CSF3R variants to identify patients at risk for progression is limited. The predictive value of RUNX1 variants appears more promising. As noted previously, the frequency of RUNX1 variants occurring in the setting of SCN-associated MDS/AML is high (68%),11 and these variants are often detectable in the blood or bone marrow months prior to progression. However, in contrast to CSF3R variants, variants in RUNX1 appear to be rare in patients with SCN without myeloid malignancy. In our study of clonal hematopoiesis in 40 patients with SCN without myeloid malignancy, none had detectable RUNX1 variants.25 Thus, RUNX1 variants may be predictive of leukemic progression in patients with SCN, although prospective data supporting this conclusion are needed. Altogether, considering the good outcomes for patients with SCN who undergo transplantation with malignancy, current evidence does not support the routine use of surveillance somatic gene panel sequencing (outside of a research study) to stratify patients for HSCT.
Timing of HSCT in SDS
One approach for early detection of clonal progression in patients with SDS is annual bone marrow surveillance. Abnormal findings on cytogenetic analysis are present in approximately 50% of patients with SDS, the most common being isochromosome 7 and del(20q).47 Isochromosome 7 results in an extra copy of the SBDS gene, which, although typically carrying a splice site variant, is capable of producing a small amount of full-length SBDS protein.48 del(20q) deletes 1 copy of EIF6, which, as discussed earlier, attenuates the defective ribosome biogenesis in SDS.49 Perhaps not surprisingly, isochromosome 7 and del(20q) are associated with a lower risk of progression to myeloid malignancy. In contrast, monosomy 7 or del(7q) is associated with rapid progression to myeloid malignancy and is an indication for transplant.50
The role for somatic gene panel sequencing in SDS is uncertain. Clonal hematopoiesis due to TP53 variants is present in approximately 50% of patients with SDS and increases with age.25 The presence of TP53-mutant clonal hematopoiesis is not associated with more severe cytopenias, and its predictive value for leukemic progression is likely to be low. Indeed, TP53 variants can persist for several years with a stable clone size without progression to myeloid malignancy. Kennedy et al20 showed that most myeloid malignancies arising in patients with SDS carry biallelic TP53 variants, raising the possibility that detection of hematopoietic clones carrying biallelic TP53 variants may be more predictive of leukemic progression. Indeed, the authors used single-cell sequencing in an informative patient with SDS to show that a subclone carrying biallelic TP53 variants was detectable 4 years prior to malignant transformation, expanded over time, and was present in the AML clone. However, single-cell sequencing is expensive, labor intensive, and not yet clinically available, limiting its current use to research applications.
CLINICAL CASES (Continued)
Case 1
This patient with ELANE-mutated SCN was noted to have a high-frequency CSF3R pathogenic variant and low-frequency DNTM3A and ASXL1 variants at age 45 years. As we have discussed, the predictive value of CSF3R variants for leukemic progression is low. Likewise, the significance of the low-frequency DNTM3A or ASXL1 variants in this older patient is unclear. We favor continued observation of this patient, reserving HSCT for the development of MDS/AML. We would not recommend repeat somatic gene panel sequencing outside of a research study. Ultimately, this patient developed AML at age 56 years, which was successfully treated by bone marrow transplantation.
Case 2
This patient with SDS presented with multiple low-abundance TP53 variants by somatic gene panel sequencing but initially had no dysplasia or chromosomal abnormality. The predictive value for leukemic progression of TP53 detected by bulk somatic DNA sequencing is likely low. It is also unknown whether, similar to patients with ARCH, the presence of multiple variants may confer a higher risk of transformation. We would favor continued observation in this patient with annual bone marrow evaluation for dysplasia and chromosomal abnormalities. The value of repeat bulk somatic gene panel sequencing to monitor TP53-mutant clone size is uncertain and not recommended outside of a research study. This patient developed AML at age 22 years, and the malignant clone demonstrated biallelic TP53 variants detectable 4 years before transformation. Although a prospective study is required, this case highlights the potential for single-cell sequencing to detect early biallelic TP53 events and serve as a biomarker for considering early HSCT.
Case 3
This 12-year-old patient with ELANE-mutant SCN was noted to have decreased G-CSF responsiveness and recurrent cellulitis. Although results of cytogenetic and somatic gene panel sequencing were negative, we would favor early HSCT in this patient. Ultimately, the patient underwent matched unrelated donor hematopoietic stem cell transplant at age 13 years and over 10 years later is alive and well without transplant-related complications.
Summary and future directions
Recent advances in our understanding of disease pathogenesis and clonal evolution have affected the clinical management of patients with congenital neutropenia. Cellular stressors play a key role in clonal evolution and depend, in part, on the underlying genetic etiology of congenital neutropenia. Thus, identifying the genetic cause of neutropenia is desirable, and we recommend panel-based testing rather than single-gene testing as part of the diagnostic evaluation. An effective surveillance strategy to identify patients at risk for transformation to myeloid malignancy is needed, particularly for patients with SDS. Surveillance strategies for patients with SCN will need to account for recent improvements in HSCT outcomes even in the presence of active myeloid malignancy.
Many open questions in the field need to be addressed and may affect clinical management of patients with congenital neutropenia. Prospective trials are needed to determine whether RUNX1 or biallelic TP53 variants are predictive of leukemic transformation in SCN or SDS, respectively. The frequency and clinical implications of clonal hematopoiesis due to chromosomal abnormalities in patients with CN are unknown. Prospective studies to detect somatic single nucleotide, small insertions/deletions, or copy number alterations in patients with CN are needed. Finally, it will be important to define risks of myeloid malignancy and patterns of clonal evolution in the expanding genetic subtypes of congenital neutropenia.
Conflict-of-interest disclosure
Julia T. Warren: no competing financial interests to declare.
Daniel C. Link: no competing financial interests to declare.
Off-label drug use
Julia T. Warren: no off-label drug use.
Daniel C. Link: no off-label drug use.
