

Abstract
The treatment landscape in acute myeloid leukemia (AML) is rapidly evolving, with multiple new therapies approved in recent years. However, the prognosis for patients with high-risk genetic subsets of AML remains poor, and the development of more effective treatment options for these patients is ongoing. Three of these high-risk AML patient subsets include TP53-mutated AML, FLT3-internal tandem duplication (ITD)-mutated AML, and AML harboring rearrangements affecting the KMT2A locus (KMT2A-r AML). The prognosis for TP53-mutated AML remains poor with both intensive and targeted regimens, including those incorporating the BCL-2 inhibitor, venetoclax. Allogeneic hematopoietic stem cell transplantation is the only potentially curative therapy for these patients, but posttransplant relapse rates remain high. Patients with FLT3-ITD-mutated AML continue to have suboptimal outcomes with standard therapies and experience high rates of relapse following transplant. KMT2A-r AML is also associated with poor outcomes with current treatment approaches, and effective standards of care are lacking for patients with relapsed/refractory disease. This article discusses current treatment approaches, along with the investigational agents being explored for the treatment of these 3 AML subsets, focusing primarily on agents that are further along in development.
Learning Objectives
Recognize the high-risk subsets of TP53-mutated, FLT3-ITD mutated, and KMT2A-rearranged AML, in which better treatment options are needed
Understand that patients with TP53-mutated AML have poor outcomes with current standard therapies and should be treated on clinical trials when possible
Review the novel FLT3 inhibitors being studied in combination with many other agents in patients with FLT3- mutated AML
Learn about the novel inhibitors of the menin-KMT2A interaction that have demonstrated promise in the treatment of KMT2A-rearranged AML
CLINICAL CASE 1
A 60-year-old man with a history of stage III colon cancer treated with surgery and adjuvant chemotherapy 8 years ago develops pancytopenia with circulating blasts. A bone marrow biopsy reveals acute myeloid leukemia (AML) with 30% myeloid blasts. Cytogenetic analysis shows a complex karyotype including monosomy 7. A next generation sequencing panel reveals a pathogenic TP53 mutation with a variant allele frequency of 30%. He is otherwise feeling well without any functional limitations and has no other notable comorbidities.
Current approaches
The TP53 protein is a transcription factor and tumor suppressor that regulates many target genes with diverse functions. The mechanism of action of TP53 along with the therapeutic agents being investigated in TP53-mutated AML is shown in Figure 1. TP53 mutations occur in 5% to 10% of de novo AML and have a higher incidence in therapy-related AML (t-AML), at 20% to 35%. TP53-mutated AML is also often associated with a complex karyotype.1,2 Most AML cases harboring TP53 derangements involve biallelic inactivation of the gene. TP53 derangements caused by mutations, deletions, and other disruptions of the 17p13 locus are associated with a very poor prognosis in AML with current approaches.
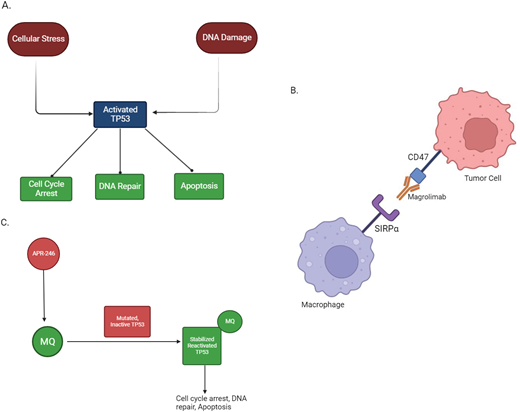
Mechanism of action of TP53 and agents under investigation for TP53-mutated AML. (A) Mechanism of action of TP53: cellular stress and DNA damage activate the TP53 protein, which leads to downstream cell cycle arrest, DNA repair, and apoptosis. Cells containing mutations affecting the TP53 protein are not able to respond to stress and DNA damage appropriately, which increases the risk of neoplastic transformation. (B) Mechanism of action of magrolimab: the binding of CD47 on tumor cells to SIRPα on macrophages inhibits phagocytosis of tumor cells. Magrolimab is a monoclonal antibody that binds to CD47 and prevents it from binding to SIRPα, which thereby facilitates tumor cell phagocytosis. (C) Mechanism of action of APR-246: APR-246 is converted to MQ under physiological conditions. MQ binds to mutated TP53 and facilitates thermodynamic stabilization of the protein, thereby shifting equilibrium to the active form of TP53. APR-246 also exhibits TP53-independent activity through glutathione depletion, which increases lipid peroxidases and other reactive oxygen species, thereby promoting cell death through ferroptosis (not depicted). Created with BioRender.com.
Mechanism of action of TP53 and agents under investigation for TP53-mutated AML. (A) Mechanism of action of TP53: cellular stress and DNA damage activate the TP53 protein, which leads to downstream cell cycle arrest, DNA repair, and apoptosis. Cells containing mutations affecting the TP53 protein are not able to respond to stress and DNA damage appropriately, which increases the risk of neoplastic transformation. (B) Mechanism of action of magrolimab: the binding of CD47 on tumor cells to SIRPα on macrophages inhibits phagocytosis of tumor cells. Magrolimab is a monoclonal antibody that binds to CD47 and prevents it from binding to SIRPα, which thereby facilitates tumor cell phagocytosis. (C) Mechanism of action of APR-246: APR-246 is converted to MQ under physiological conditions. MQ binds to mutated TP53 and facilitates thermodynamic stabilization of the protein, thereby shifting equilibrium to the active form of TP53. APR-246 also exhibits TP53-independent activity through glutathione depletion, which increases lipid peroxidases and other reactive oxygen species, thereby promoting cell death through ferroptosis (not depicted). Created with BioRender.com.
For patients treated with intensive chemotherapy, complete remission (CR) rates are less than 30%, and median overall survival (OS) is less than 1 year.1,3 For patients who respond to induction therapy, subsequent allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only potentially curative treatment. However, posttransplant outcomes also remain poor, with a median posttransplant OS of less than 1 year and a 2-year OS rate of less than 30%.4,5
There has been interest in less intensive treatments, including hypomethylating agents (HMAs), in this patient population given the poor prognosis associated with intensive chemotherapy. One single-arm study found that 10-day decitabine therapy was associated with favorable response rates in TP53-mutated AML.6 However, this study included only 21 patients with TP53 mutations. Other studies have found poor OS of less than 1 year in patients with TP53 mutations treated with azacitidine, 5-day decitabine, and 10-day decitabine.7,8 One of these was a randomized study involving 71 patients, 30% of whom had TP53 mutations, that compared 5-day vs 10-day decitabine. Among patients with TP53 mutations, there was no difference in response rates with 5-day vs 10-day decitabine. Median OS also did not differ significantly and was less than 6 months in both groups, indicating no advantage of 10-day over 5-day decitabine.
The addition of venetoclax, a BCL-2 inhibitor, to HMA therapy has, unfortunately, not shown the improvement in patient outcomes to the level seen in other genetic subtypes of AML. Among patients with TP53-mutated disease treated in the phase 3 VIALE-A study, the combination of azacitidine-venetoclax yielded an encouraging CR with an incomplete hematologic recovery (CRi) rate of 55% (95% CI, 38.3-71.4) compared to 0% in patients treated with azacitidine plus placebo (P < .001).9 However, a retrospective review of patients with TP53-mutated AML and poor-risk cytogenetics treated both in VIALE-A and the phase 1b study preceding VIALE-A found that the combination of venetoclax plus an HMA did not translate into improved duration of response or OS compared to an HMA alone.10 Another study of 10-day decitabine plus venetoclax in patients with newly diagnosed AML found that OS was significantly worse in TP53- mutated vs wild-type patients, with a median OS of less than 6 months in patients with TP53 mutations.11 These results further indicate the lack of improvement with 10-day decitabine with or without venetoclax in this patient population. The overall poor outcomes with the current available standards of care highlight the need to develop additional novel therapies beyond venetoclax to improve survival and long-term outcomes in patients with TP53-mutated AML.
Targeting CD47 in TP53-mutated AML
The CD47 protein is overexpressed in myeloid malignancies, including in AML leukemic stem cells. The CD47 protein acts as a “do not eat me” signal that inhibits phagocytosis of tumor cells by binding to the signal regulatory protein alpha on macrophages. In preclinical studies, CD47 blockade overcomes this inhibition and stimulates tumor cell killing by macrophages (Figure 1B).12 A phase 1b study combining the CD47 antibody magrolimab with azacitidine for the treatment of patients with newly diagnosed AML ineligible for intensive chemotherapy showed promising efficacy in both TP53-mutated and TP53-wild-type patients. The objective response rate was 65% for all comers and 71% for TP53-mutated patients. Median OS was 18.9 months for TP53-wild-type patients and 12.9 months for TP53-mutated patients.13 A triplet combination of magrolimab-azacitidine- venetoclax was subsequently studied in a phase 1b/2 clinical trial. In patients with newly diagnosed AML, the CR/CRi rate was 94% for all patients and 100% for patients with TP53-mutated disease.14 The most common grade 3/4 adverse events were pneumonia, febrile neutropenia, hyperbilirubinemia, elevated alanine aminotransferase, and skin infection. CD47 is expressed on red blood cells, and it is common to see a drop in hemoglobin, particularly early in the course of treatment. The ongoing phase 3 trials Enhance-2 (NCT04778397) and Enhance-3 (NCT05079230) are further addressing the role of magrolimab in TP53-mutated and TP53-wild-type disease, respectively. Other CD47 antibodies are currently in development in earlier-phase clinical trials.
TP53 stabilization in TP53-mutated AML
APR-246 (epranetapopt) acts as a TP53 stabilizer and is a prodrug that converts to methylene quinuclidinone (MQ) under physiological conditions. MQ binds to cysteine residues in mutant TP53, which leads to thermodynamic stabilization of the protein and shifts equilibrium toward the functional conformation (Figure 1C).15 APR-246 has also been shown to deplete glutathione. Glutathione depletion increases lipid peroxidases and other reactive oxygen species, which increases susceptibility to ferroptosis.16 Ferroptosis induction has been shown to be an important mechanism of the early leukemic cell death induced by APR-246 regardless of TP53 mutation status.17 A phase 1b/2 study of APR-246–azacitidine in patients with TP53-mutated myelodysplastic syndromes (MDS) or oligoblastic AML (20%-30% blasts) found an overall response rate (ORR) of 71%, with 44% of patients achieving CR.18 Despite the promising results, a subsequent phase 3 trial (NCT03745716) failed to meet its primary end point of a superior CR rate with APR-246–azacitidine compared to azacitidine alone in patients with TP53-mutated MDS. However, a subsequent phase 2 study of APR-246–azacitidine as maintenance therapy following allo-HSCT in patients with TP53-mutated AML or MDS showed promising results compared to historical data, with a median relapse-free survival of 368 days and a median OS of 586 days.19 Another phase 1 study of the triplet combination of APR-246–venetoclax– azacitidine for the primary treatment of TP53-mutated AML showed a CR rate of 37% and a CR/CRi rate of 53%, meeting the Simon's 2-stage efficacy criteria.20
CLINICAL CASE 1 (Continued)
The patient presented here is a fit 60-year-old man with TP53- mutated, therapy-related AML and minimal comorbidities. An up-front discussion regarding the limited treatment options and poor prognosis is very important for shared decision-making that aligns with the patient's treatment goals. If possible, he should be referred for a clinical trial focusing on TP53-mutated AML. A summary of our approach for the treatment of newly diagnosed TP53-mutated AML is presented in Figure 2.
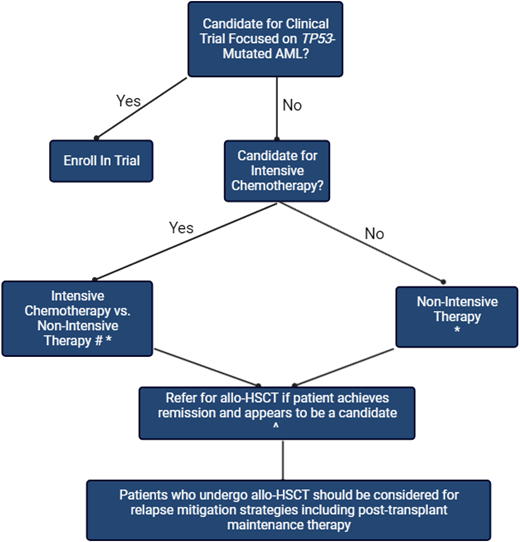
Treatment approach for newly diagnosed TP53-mutated AML.
# Many clinicians prefer less intensive regimens to reduce toxicity, even in patients who are eligible, given the poor outcomes associated with intensive chemotherapy. Robust data supporting this practice are lacking.
* Adding venetoclax to the treatment regimen may improve the likelihood of response, but mounting data suggest that this is unlikely to improve survival.
^ Even though allo-HSCT is the only potentially curative therapy, posttransplant outcomes remain poor. Consequently, the risk/benefit assessment ends up not favoring allo-HSCT for many patients.
Created with BioRender.com.
Treatment approach for newly diagnosed TP53-mutated AML.
# Many clinicians prefer less intensive regimens to reduce toxicity, even in patients who are eligible, given the poor outcomes associated with intensive chemotherapy. Robust data supporting this practice are lacking.
* Adding venetoclax to the treatment regimen may improve the likelihood of response, but mounting data suggest that this is unlikely to improve survival.
^ Even though allo-HSCT is the only potentially curative therapy, posttransplant outcomes remain poor. Consequently, the risk/benefit assessment ends up not favoring allo-HSCT for many patients.
Created with BioRender.com.
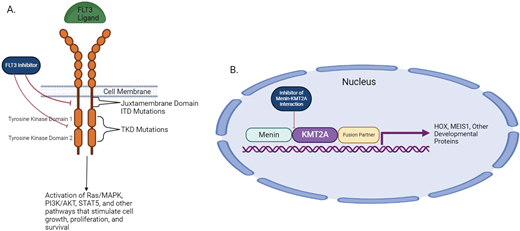
Pathogenesis of FLT3-mutated AML and KMT2A-r AML. (A) FLT3-ITD and FLT3-TKD mutations lead to increased downstream signaling from the FLT3 receptor, which thereby promotes increased cell growth, proliferation, and survival. FLT3 inhibitors block this downstream signaling. (B) Binding of the KMT2A protein to menin allows the KMT2A/fusion partner protein to facilitate downstream transcription of HOX and other developmental proteins that contribute to leukemic pathogenesis. Inhibitors of the menin- KMT2A interaction inhibit this transcription. AKT, Akt serine/threonine protein kinases; MAPK, mitogen-activated protein kinases; PI3K, phosphoinositide-3 kinases; STAT5, signal transducer and activator of transcription 5. Created with BioRender.com.
Pathogenesis of FLT3-mutated AML and KMT2A-r AML. (A) FLT3-ITD and FLT3-TKD mutations lead to increased downstream signaling from the FLT3 receptor, which thereby promotes increased cell growth, proliferation, and survival. FLT3 inhibitors block this downstream signaling. (B) Binding of the KMT2A protein to menin allows the KMT2A/fusion partner protein to facilitate downstream transcription of HOX and other developmental proteins that contribute to leukemic pathogenesis. Inhibitors of the menin- KMT2A interaction inhibit this transcription. AKT, Akt serine/threonine protein kinases; MAPK, mitogen-activated protein kinases; PI3K, phosphoinositide-3 kinases; STAT5, signal transducer and activator of transcription 5. Created with BioRender.com.
CLINICAL CASE 2
A 50-year-old man with no major comorbidities and good performance status presents to discuss treatment options for AML diagnosed in the context of fatigue and circulating blasts. A bone marrow biopsy demonstrates 80% blasts. Polymerase chain reaction with capillary electrophoresis for FLT3 mutations detects the presence of an FLT3 internal tandem duplication (ITD) mutation. The karyotype is normal, and no other mutations are detected on next generation sequencing.
Background
Mutations in the FMS-like tyrosine kinase 3 (FLT3) protein are present in approximately 30% of newly diagnosed AML cases. These mutations are classified as ITD mutations affecting the juxtamembrane domain or point mutations in the tyrosine kinase domain (TKD mutations). FLT3-ITD mutations have historically been established as a poor prognostic factor.21 FLT3-TKD mutations have not demonstrated a consistent prognostic impact.22 The 2017 European LeukemiaNet prognostic classification system separated FLT3-ITD-mutated AML into different categories based on the ITD allelic ratio and the presence of a concurrent NPM1 mutation.23 However, the updated 2022 ELN classification includes all FLT3-ITD-mutated AML in the intermediate risk category.24 The current standard of care for most patients with FLT3-ITD mutations who are candidates is to undergo allo-HSCT in first CR. However, patients with FLT3-ITD mutations are also at increased risk of relapse following transplant.25 Recent efforts in FLT3-mutated AML have focused on developing novel FLT3 inhibitors, combining FLT3 inhibitors with other therapies to overcome therapeutic resistance, expanding treatment options for patients with relapsed/refractory (R/R) disease, and studying posttransplant maintenance therapies to reduce the risk of posttransplant relapse. The pathogenesis of FLT3-mutated AML and the mechanism of action of FLT3 inhibitors is shown in Figure 3A.

Treatment recommendations for FLT3-Mutated AML. Color code: Blue background with white text: standard of care; green background with red text: investigational approaches. Aza, azacitidine; ELN, European Leukemia Network; Gilt, gilteritinib; IC, intensive chemotherapy; LDAC, low dose cytarabine; ND, newly diagnosed; R/R, relapsed/refractory; Ven, venetoclax.
*Data exist supporting allo-HSCT for all patients with FLT3-ITD-mutated AML, even if they are classified as favorable risk by the 2017 ELN guidelines based on a low ITD allelic ratio and concurrent NPM1 mutation. The updated 2022 ELN guidelines classify all FLT3-ITD-mutated AML as intermediate risk.
**Sorafenib maintenance after allo-HSCT is approved per National Comprehensive Cancer Network guidelines but not commonly used in clinical practice. Sorafenib is only effective against FLT3-ITD mutations, not FLT3-TKD mutations.
Created with BioRender.com.
Treatment recommendations for FLT3-Mutated AML. Color code: Blue background with white text: standard of care; green background with red text: investigational approaches. Aza, azacitidine; ELN, European Leukemia Network; Gilt, gilteritinib; IC, intensive chemotherapy; LDAC, low dose cytarabine; ND, newly diagnosed; R/R, relapsed/refractory; Ven, venetoclax.
*Data exist supporting allo-HSCT for all patients with FLT3-ITD-mutated AML, even if they are classified as favorable risk by the 2017 ELN guidelines based on a low ITD allelic ratio and concurrent NPM1 mutation. The updated 2022 ELN guidelines classify all FLT3-ITD-mutated AML as intermediate risk.
**Sorafenib maintenance after allo-HSCT is approved per National Comprehensive Cancer Network guidelines but not commonly used in clinical practice. Sorafenib is only effective against FLT3-ITD mutations, not FLT3-TKD mutations.
Created with BioRender.com.
Current approaches
The current standard frontline therapy for patients with newly diagnosed FLT3-mutated AML who are eligible for intensive chemotherapy is induction therapy with midostaurin combined with traditional “7 + 3” followed by consolidation with midostaurin- cytarabine. Midostaurin is a first-generation type 1 FLT3 inhibitor with activity against both ITD- and TKD-mutated receptors. This approach was informed by the phase 3 RATIFY trial in which midostaurin outperformed placebo when combined with chemotherapy in patients aged 18 to 59 (hazard ratio for death, 0.78; one-sided P = .009).26 Though the RATIFY study only included patients aged 18 to 59, there is no age limitation on the US Food and Drug Administration's approval.
For patients who are ineligible for intensive chemotherapy, the combination of venetoclax plus HMA therapy can be given frontline. The composite CR rate was 72.4% in patients with FLT3-mutated AML treated with frontline venetoclax-azacitidine on the VIALE-A trial.9
The current standard of care for patients with R/R FLT3- mutated AML is monotherapy with the second-generation type 1 FLT3 inhibitor gilteritinib. This practice was based on results of the phase 3 Admiral trial, in which gilteritinib was found to be superior to standard of care chemotherapy in this setting (median OS, 9.3 months vs 5.6 months; hazard ratio, 0.64; 95% CI, 0.49-0.83; P < .001).27 For patients with FLT3-ITD-mutated AML who undergo allo-HSCT, sorafenib (a type 2 FLT3 inhibitor active against ITD- mutated but not TKD-mutated AML) is sometimes given off-label as posttransplant maintenance therapy based on the results of 2 clinical trials, one of which was placebo controlled.28,29
Investigational approaches
Intensive chemotherapy candidates
Several clinical trials are ongoing involving gilteritinib in patients with FLT3-mutated AML, many of which combine gilteritinib with intensive chemotherapy. A phase 1 study combining gilteritinib with 7 + 3 was well tolerated and associated with a FLT3 mutation clearance rate of 70%.30 Additional FLT3 inhibitors are also under investigation. Quizartinib is a second- generation type 2 inhibitor that has been compared to placebo in combination with 7 + 3 in patients with newly diagnosed, FLT3-ITD-mutated AML in the Quantum First trial (NCT02668653). At a median follow-up of 39.2 months, OS was significantly longer in the quizartinib arm (31.9 months) vs the placebo arm (15.1 months; hazard ratio, 0.776; 95% CI, 0.615-0.979; 2-sided P = .0324). CR/CRi rates were 71.6% in the quizartinib arm vs 64.9% in the placebo arm.31 It is notable that this trial included patients aged 18 to 75 compared to the RATIFY trial, which had an upper age cutoff of 59. Crenolanib is a second- generation, type 1 FLT3 inhibitor that has also demonstrated promising results in early-phase trials.32,33 FLT3 inhibitors are also being investigated as maintenance therapy post transplant for patients undergoing allo-HSCT. Posttransplant midostaurin was found to be associated with superior event-free survival compared to a historic control in 1 study.34 Another study found that posttransplant midostaurin improved relapse-free survival compared to standard of care only when FLT3 phosphorylation was inhibited to less than 70% of baseline.35 Gilteritinib is also being compared to placebo as maintenance therapy following allo-HSCT through the Blood and Marrow Transplant Clinical Trials Network Protocol 1506 (NCT02997202) and following chemotherapy in patients who are not undergoing allo-HSCT (NCT02927262).
Nonintensive chemotherapy candidates
For those patients who are not candidates for intensive chemotherapy, gilteritinib is also being studied in combination with HMA therapy and other targeted therapies. The recently completed phase 3 LACEWING trial compared the combination of gilteritinib-azacitidine to azacitidine alone in patients with newly diagnosed FLT3-mutated AML who were ineligible for intensive chemotherapy. The combination arm yielded superior response rates but not superior OS, and the study therefore did not meet its primary end point.36
There is evidence that BCL-2 upregulation may play a role in resistance to FLT3 inhibition and that adding venetoclax may help to overcome this.37 A phase 1b study of gilteritinib-venetoclax in patients with R/R disease found a favorable ORR of 90% in patients with FLT3-mutated disease.38 An ongoing phase 1/2 study (NCT04140487) of gilteritinib-venetoclax-azacitidine found an ORR of 67% with a median OS of 10.5 months at a median follow-up of 9.9 months in patients with R/R disease. The ORR was 100% in newly diagnosed patients ineligible for chemotherapy, with median OS not reached at a median follow-up of 3.8 months.39 Another study of venetoclax-decitabine-FLT3 inhibitor of investigator's choice found an ORR of 62% with 2-year OS of 29% in patients with R/R disease and an ORR of 92% with 2-year OS of 80% in patients over 60 years of age with newly diagnosed disease.40
CLINICAL CASE 2 (Continued)
The patient presented in this case has AML harboring an FLT3-ITD mutation and is an intensive induction candidate. Again, it is important to determine the patient's underlying treatment goals through shared decision-making. A summary of our approach and investigational strategies for the treatment of FLT3-mutated AML, both newly diagnosed and R/R, is presented in Figure 4.
CLINICAL CASE 3
A 65-year-old woman without major comorbidities and normal performance status is diagnosed with AML harboring a cytogenetic translocation involving 11q23. She is treated with frontline venetoclax-azacitidine and achieves CR. However, 4 months later she is noted to have relapsed AML still harboring the 11q23 translocation.
Background
The KMT2A gene (previously known as MLL1) codes for the histone-lysine N-methyltransferase 2A protein and is located on chromosome 11q23. Translocations involving the KMT2A locus occur in approximately 3% of AML cases and have a negative impact on prognosis.23,41 Translocations involving the KMT2A locus result in the expression of fusion proteins that enable an aberrant transcription program characterized by the overexpression of HOX and other developmental genes.42,43 The binding of these fusion proteins to menin, a scaffold protein, leads to the nuclear localization of the fusion proteins, thereby facilitating the aberrant transcription that is central to the pathogenesis of KMT2A-rearranged (KMT2A-r) AML.44,45 The interaction of menin with the wild-type KMT2A protein has also been shown to play a role in the pathogenesis of NPM1-mutated AML.46
Targeting menin inhibition in KMT2A-rearranged AML
SNDX-5613 is an oral small-molecule inhibitor that binds with high affinity to the KMT2A-binding pocket of menin. This inhibits the interaction between menin and KMT2A, which thereby inhibits oncogenic expression and cellular proliferation. SNDX-5613 has demonstrated activity against KMT2A-r and NPM1- mutated leukemia in preclinical and xenograft models. Augment-1 was the first-in-human phase 1 study involving SNDX-5613 and included patients with R/R KMT2A-r and NPM1-mutated acute leukemias (both acute lymphocytic leukemia and AML). This study demonstrated promising response rates, with a composite CR of 44% and a median duration of response of 5.2 months. The only dose-limiting toxicity was grade 3 QTc prolongation, which occurred in 8% of patients, all of whom were clinically asymptomatic. Other common treatment-related adverse events were nausea (22%), vomiting (17%), differentiation syndrome (DS; 15%), and diarrhea (11%).47 A follow-up phase 2 study is ongoing. SNDX-5613 is also being studied in combination with intensive chemotherapy in a phase 1 trial in patients with R/R AML or acute lymphocytic leukemia harboring a KMT2A rearrangement or NPM1 mutation (NCT05326516). An all-oral triplet regimen of SNDX-5613–ASTX727–venetoclax is being studied in a phase 1/2 clinical trial in patients with R/R AML or mixed phenotype acute leukemia (NCT05360160). The Beat AML Master Clinical trial is also studying the combination of SNDX-5613–azacitidine–venetoclax in patients older than 60 with newly diagnosed KMT2A-r or NPM1-mutated AML (NCT03013998).
KO-539 and JNJ-75276617 are 2 other novel inhibitors of the menin-KMT2A interaction that are being investigated in early- phase clinical trials in patients with R/R AML (NCT04067336, NCT04811560). DS is an important adverse event for clinicians to be aware of and, if unrecognized, can be fatal. DS was typically low grade in patients treated on Augment-1 but did lead to the US Food and Drug Administration placing a temporary hold on KO-539, which has since been lifted. Signs and symptoms of DS include unexplained fever, weight gain, edema, pleural or pericardial effusions, radiographic opacities, dyspnea, hypotension, renal dysfunction, rash, and/or a rapidly increasing white blood cell count. Treatment involves the prompt initiation of steroids and following the investigator brochure guidelines regarding continuation of the investigational agent. The pathogenesis of KMT2A-r AML is shown in Figure 3B.
CLINICAL CASE 3 (Continued)
Patients with newly diagnosed KMT2A-r AML who are candidates should undergo allo-HSCT if they achieve remission with frontline therapy. There is no current standard of care for R/R disease, but investigational inhibitors of the menin-KMT2A interaction represent promise in this setting.
Conclusions
TP53 mutations, FLT3-ITD mutations, and KMT2A rearrangements represent high-risk genetic features in AML, and improved outcomes are needed in these AML patient subsets. Multiple novel agents and therapeutic combinations are being studied in these AML subsets that have the potential to foster these needed improvements. This is an exciting time in AML research, with hope for better outcomes in patients with high-risk disease moving forward.
Conflict-of-interest disclosure
Kieran D. Sahasrabudhe: no competing financial interests to declare.
Alice S. Mims: scientific advisory committee: Syndax Pharmaceuticals, AbbVie, Genentech, BMS, Astellas, Servier Pharmaceuticals, Ryvu Therapeutics; data and safety monitoring committee: Jazz Pharmaceuticals, Daiichi Saynko; senior medical director: Leukemia and Lymphoma Society Beat AML study.
Off-label drug use
Kieran D. Sahasrabudhe: the off-label use of sorafenib is discussed.
Alice S. Mims: the off-label use of sorafenib is discussed.
