

Abstract
The BCR-ABL-negative myeloproliferative neoplasms (MPNs) have a variable risk of progressing to accelerated- or blast-phase MPN (MPN-AP/MPN-BP), defined by the presence of 10% to 19% and more than or equal to 20% myeloid blasts in the peripheral blood or bone marrow, respectively. The molecular processes underlying the progression to MPN-AP/MPN-BP are becoming increasingly understood with the acquisition of additional mutations in epigenetic modifiers (eg, ASXL1, EZH2, TET2), TP53, the Ras pathway, or splicing factors (eg, SRSF2, U2AF1), having been described as important steps in this evolutionary process. At least partially driven by the enrichment of these high-risk molecular features, the prognosis of patients with MPN-BP remains inferior to other patients with acute myeloid leukemia, with a median overall survival of 3 to 6 months. Allogeneic hematopoietic cell transplantation remains the only potentially curative therapeutic modality, but only a minority of patients are eligible. In the absence of curative intent, therapeutic strategies or palliative treatment with hypomethylating agents as monotherapy or in combination with ruxolitinib or venetoclax can be considered. Several novel agents are in various stages of clinical development but are not available for routine use at this point, highlighting the need for ongoing research and the prioritization of clinical trial enrollment when feasible.
Learning Objectives
Understand the definition, clinical characteristics, and prognosis of patients with MPN-AP or MPN-BP
Review the molecular evolution underlying the progression from chronic-phase MPN to MPN-AP or MPN-BP
Discuss current treatment options for MPN-AP and MPN-BP, including allogeneic hematopoietic cell transplantation, intensive chemotherapy, hypomethylating agents, emerging combinations, and molecularly targeted therapies
CLINICAL CASE
A 65-year-old male with a past medical history of atherosclerotic cardiovascular disease with myocardial infarction and coronary artery bypass grafting was referred to hematology for further workup of thrombocytosis, with a platelet count of 933 × 109/L found on routine blood work. The remainder of his laboratory test results included a normal white blood cell count of 8.3 × 109/L with normal differential; a hemoglobin level of 13.5 g/dL; normal electrolytes, kidney, and liver function; and an elevated lactate dehydrogenase level (352 U/L; normal range, 120-246 U/L). There was no evidence of leukoerythroblastic changes on peripheral blood smear review. A computed tomography scan of the abdomen and pelvis demonstrated a normal spleen size. Molecular testing from the peripheral blood was positive for a JAK2 V617F mutation, and the patient was diagnosed with essential thrombocythemia (ET). A bone marrow biopsy and aspirate was performed that was consistent with ET. Given his age and cardiovascular comorbidity, he received cytoreductive therapy with hydroxyurea as well as aspirin. Five years after the initial diagnosis, the patient developed worsening anemia requiring red blood cell transfusions. Bone marrow biopsy showed a markedly hypercellular marrow with profound myeloid hyperplasia, hypoplastic erythropoiesis, markedly hypoplastic megakaryopoiesis, marked fibrosis and osteosclerosis, and no increase in blasts, consistent with progression to post-ET myelofibrosis. Over the subsequent 5 months, the patient developed progressive leukocytosis, worsening red blood cell transfusion requirements, and thrombocytopenia. A bone marrow biopsy was repeated and was consistent with acute myeloid leukemia (AML) with 83% blasts.
Introduction
The BCR-ABL1-negative myeloproliferative neoplasms (MPNs) compose a heterogeneous spectrum of diseases with a variable risk of progression to accelerated-phase MPN (MPN-AP) and blast-phase MPN (MPN-BP), which is defined as an increase in myeloid blasts in the peripheral blood or bone marrow to 10% to 19% and greater than or equal to 20%, respectively.1-3 While patients with chronic-phase MPN present with constitutional symptoms, thromboembolic and hemorrhagic complications, splenomegaly and variable degrees of abnormal peripheral blood counts, patients with MPN-AP or MPN-BP typically present with progressive anemia, thrombocytopenia, and leukocytosis and have a poor prognosis with a median overall survival (OS) of only 3 to 6 months in retrospective cohort studies.4-6 Although a better understanding of the molecular landscape of MPN has led to the development and approval of several agents for chronic-phase MPN, such as the Jak inhibitors ruxolitinib, fedratinib, and pacritinib for myelofibrosis as well as ropeg-interferon (IFN) for polycythemia vera (PV), no current standard of care for MPN-AP and MPN-BP exists.7-10 However, thanks to advances in molecular testing, the pathophysiology underlying the progression of chronic-phase MPN into MPN-AP and MPN-BP is becoming increasingly characterized, and several clinical trials of novel combination therapies have recently been conducted. Herein, we review the current risk assessment for patients with chronic- phase MPNs with regard to predicting disease progression, recent developments in understanding the clonal evolution driving MPN progression, and emerging treatment options.
Risk prediction and genetic landscape of MPN-BP
While the majority of patients with chronic-phase MPNs do not experience disease progression, this cumulative lifetime risk is variable for patients with ET, PV, and primary myelofibrosis (PMF) and ranged from 3.8% for ET patients to 14.2% for PMF patients in a large cohort study of patients treated at the Mayo Clinic.11 As the only potentially curative therapeutic modality for MPNs remains allogeneic hematopoietic cell transplant (allo-HCT), risk-stratification tools allowing for the appropriate triaging and referral of patients for allo-HCT are of great clinical relevance.12,13 Various prognostic scoring systems for both PMF (eg, the dynamic prognostic scoring system) as well as post-ET and post-PV MF (myelofibrosis secondary to PV and ET Collaboration Prognostic Model) have been developed to predict OS but also correlate, albeit imperfectly, with the risk of progression to MPN-BP.14-16 More recently, the addition of selected high-risk mutations (ASXL1, EZH2, SRSF2, IDH1/2) and the absence of a type 1 CALR mutation to other prognostic disease and clinical characteristics has led to the development of the Molecularly Inspired Prognostic Scoring System (MIPSS).15 Conversely, the genetically inspired scoring system is based only on molecular features such as karyotype and selected high-risk mutations.17 Both scores have been shown to predict prognosis in PMF patients.15,17
Table 1 provides an overview of prognostic factors associated with an increased risk of progression to MPN-BP. These can be classified into disease related (eg, increased peripheral blood blasts, higher-grade bone marrow fibrosis, depth of cytopenias), molecular (eg, the presence of certain high-risk somatic mutations), and treatment related.2,16,18 While the leukemogenic potential of hydroxyurea remains under evaluation, other agents used for cytoreduction in the past, such as busulfan and pipobroman, have been associated with an increased risk of progression to MPN-BP. Conversely, treatment with IFN may be associated with a reduced risk of progression to MPN-BP.16,19,20
Patient, disease, and treatment characteristics associated with risk of progression to MPN-AP/MPN-BP
| Patient factor |
| Advanced patient age (>60 vs >70 years)57,58 |
| Disease factors |
| Longer disease duration31 |
| Higher risk for patients with PMF compared to ET and PV11 |
| Platelet count <150 × 109/L, leukocytosis, transfusion dependence, peripheral blasts ≥3%16,58,59 |
| Splenomegaly of ≥20 cm from left costal margin or requirement for splenectomy59 |
| Bone marrow reticulin fibrosis57 |
| Abnormal karyotype58,59 |
| Lower risk with CALR mutations60 |
| High-risk mutations (eg, TP53, ASXL1, SRSF2, IDH2, EZH2)4,60 |
| Higher DIPSS, MYSEC-PM, or MIPSS70v2.0 score16 |
| Treatment factors |
| Prior treatment with alkylating agents and pipobroman20,58 |
| Lower risk in patients treated with IFN16 |
| Patient factor |
| Advanced patient age (>60 vs >70 years)57,58 |
| Disease factors |
| Longer disease duration31 |
| Higher risk for patients with PMF compared to ET and PV11 |
| Platelet count <150 × 109/L, leukocytosis, transfusion dependence, peripheral blasts ≥3%16,58,59 |
| Splenomegaly of ≥20 cm from left costal margin or requirement for splenectomy59 |
| Bone marrow reticulin fibrosis57 |
| Abnormal karyotype58,59 |
| Lower risk with CALR mutations60 |
| High-risk mutations (eg, TP53, ASXL1, SRSF2, IDH2, EZH2)4,60 |
| Higher DIPSS, MYSEC-PM, or MIPSS70v2.0 score16 |
| Treatment factors |
| Prior treatment with alkylating agents and pipobroman20,58 |
| Lower risk in patients treated with IFN16 |
DIPSS, Dynamic International Prognostic Scoring System; MYSEC-PM, myelofibrosis secondary to PV and ET Collaboration Prognostic Model.
Significant advances have been made in elucidating the genetic landscape of MPN-BP. In addition to the mutations in JAK2, CALR, and MPL that are present in approximately 90% of patients with chronic-phase MPN, patients with MPN-BP have been shown to harbor additional somatic mutations, with ASXL1, TET2, TP53, EZH2, RUNX1, and splicing factor genes (eg, SRSF2, U2AF1) being the most frequently encountered genetic alterations.18,21-23 This mutational profile of MPN-BP is distinct from patients with de novo AML, and the enrichment of high-risk mutations might be contributing to the adverse prognosis of patients with MPN-BP.24 For example, mutations in FLT3 occur in up to 30% of AML cases but are rare in MPN-BP, which is characterized by a higher frequency of mutations in ASXL1, TET2, TP53, IDH1/2, and splicing factor genes (eg, SRSF2, U2AF1).18,21-23,25 Serial assessment of the mutational profile of patients progressing from chronic to MPN-AP and MPN-BP have demonstrated that the serial acquisition of additional mutations—for example, in IDH1/2 or TET2—as well as the expansion of a previously present subclone harboring a TP53 mutation can potentially drive disease progression.22,26-30 However, mutations in other genes such as ASXL1 or EZH2 can be acquired either before or after the canonical driver mutations in JAK2, MPL, or CALR.26 Additionally, recent studies have highlighted the contribution of a pro-inflammatory microenvironment in the bone marrow as a potential driver of clonal evolution and disease progression.30-32 Despite these advances, additional research is needed to understand the clonal evolution among patients undergoing various forms of treatment and whether serial sequencing can be used to predict transformation and inform treatment recommendations (eg, referral for allo-HCT).
Treatment of MPN-AP and MPN-BP
A proposed treatment approach to MPN-BP is outlined in Figure 1. As the prognosis of patients with MPN-BP is limited and has not substantially improved over the last decade, clinical trial enrollment is encouraged wherever feasible.4 The only potentially curative therapeutic modality remains allo-HCT, which should be strongly considered for eligible patients with MPN-BP. Outcomes of allo-HCT in patients with MPN-BP are variable but appear to be inferior compared to patients with other subtypes of AML in large registry studies such as a recent analysis from the Center for International Blood and Marrow Transplant Research, which reported a hazard ratio (HR) of death of 1.4 (95% CI, 1.12-1.76) even among patients in remission at the time of transplant.33 This appears to be primarily driven by a higher rate of relapse and not by differences in nonrelapse mortality for MPN-BP compared to de novo or post-MDS AML.34 Interestingly, there was no statistically significant difference among patients with active disease at the time of allo-HCT and those who received myeloablative or reduced-intensity conditioning.33 Other studies have reported a 3-year OS of 18% to 33% for MPN-BP patients undergoing allo-HCT.35,36 Emerging data suggest that patients with TP53 mutations have inferior outcomes after allo-HCT, with TP53-mutant patients having a higher risk of death (HR, 1.99; 95% CI, 1.14-3.49) and relapse (HR, 2.59; 95% CI, 1.41-4.74).34 Given that disease relapse appears to be a main driver of mortality after allo-HCT among MPN-BP patients, efforts to reduce the risk of relapse are of great interest. Other areas of ongoing research include the influence of conditioning regimen intensity, graft source, and the influence of molecular characteristics on posttransplant outcomes.4,33,34
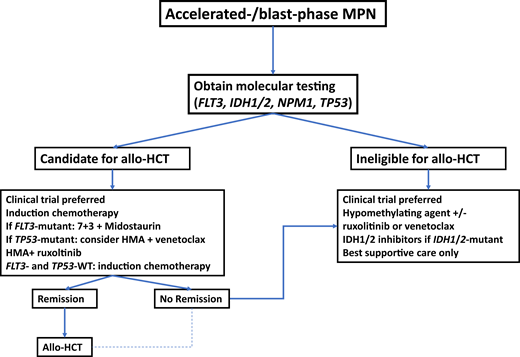
Potential treatment algorithm for patients with blast-phase MPN.
In the absence of high-quality prospective studies specific to patients with blast-phase MPN, treatment recommendations are extrapolated from studies of patients with AML. At the time of diagnosis, molecular testing should be obtained. In patients who are potential candidates for allo-HCT, this constitutes the only potentially curative therapeutic modality and should therefore be considered. Induction chemotherapy prior to transplant is informed by molecular disease features, with the addition of midostaurin in patients with FLT3 mutations. Due to lower response rates with conventional cytotoxic chemotherapy in patients with TP53 mutations, combination therapy with HMAs plus venetoclax could be considered. Patients in remission after induction chemotherapy should proceed to allo-HCT if eligible. which may also be an option for a selected subset of patients who do not achieve a complete remission with induction therapy. For transplant-ineligible patients and those with R/R disease, clinical trial enrollment, HMAs alone or in combination, IDH1/2 inhibitors, or best supportive care are therapeutic options.
Potential treatment algorithm for patients with blast-phase MPN.
In the absence of high-quality prospective studies specific to patients with blast-phase MPN, treatment recommendations are extrapolated from studies of patients with AML. At the time of diagnosis, molecular testing should be obtained. In patients who are potential candidates for allo-HCT, this constitutes the only potentially curative therapeutic modality and should therefore be considered. Induction chemotherapy prior to transplant is informed by molecular disease features, with the addition of midostaurin in patients with FLT3 mutations. Due to lower response rates with conventional cytotoxic chemotherapy in patients with TP53 mutations, combination therapy with HMAs plus venetoclax could be considered. Patients in remission after induction chemotherapy should proceed to allo-HCT if eligible. which may also be an option for a selected subset of patients who do not achieve a complete remission with induction therapy. For transplant-ineligible patients and those with R/R disease, clinical trial enrollment, HMAs alone or in combination, IDH1/2 inhibitors, or best supportive care are therapeutic options.
Since outcomes of patients undergoing allo-HCT with active disease are potentially worse compared to patients in remission at the time of transplant,35,36 initial treatment at the time of disease progression is an important consideration and area of ongoing research. Selected clinical trials are summarized in Table 2. For patients eligible for intensive chemotherapy, such as an anthracycline-cytarabine–based regimen, response rates of 30% to 60% have been reported, with a median OS of only 3 to 9 months among patients who did not subsequently undergo an allo-HCT despite achieving a response to initial treatment.4,6,37 Patients who were able to proceed to allo-HCT had a significantly longer OS compared to patients who did not receive transplants.4,6,37 Furthermore, the survival of patients who receive intensive chemotherapy but do not receive consolidation with allo-HCT does not appear to differ from patients treated with nonintensive approaches.37 As such, the use of intensive chemotherapy should only be considered in patients planned or eligible to receive allo-HCT.
Selected studies of treatment outcomes in MPN-BP
| Therapeutic regimen | Patient population | Study design | Key outcomes | Reference |
|---|---|---|---|---|
| Intensive chemotherapy | ||||
| Intensive chemotherapy (not specified) | MPN-BP | Retrospective cohort study | Median OS 10.2 mo | 61 |
| “7 + 3” cytarabine plus an anthracycline | MPN-BP | Retrospective cohort study | CR rate 67%; median OS 19.2 mo for allo-HCT vs 3.8 mo for nontransplant patients | 5 |
| AML-like induction chemotherapy | MPN-BP | Retrospective cohort study | CR rate 35%; 3-y OS 32% for allo-HCT vs 19% for nontransplant patients | 4 |
| HMA monotherapy | ||||
| Azacitidine | MPN-BP/AP | Retrospective cohort study | ORR 61.5%; median OS 13.5 mo | 43 |
| MPN-BP | Retrospective cohort study | ORR 68%; median OS 9.9 mo | 42 | |
| MPN-BP | Retrospective cohort study | Median OS 9 mo | 61 | |
| Azacitidine or decitabine | MPN-BP | Retrospective cohort study | Median OS 6.7 mo | 5 |
| Decitabine | MPN-BP, MPN-AP, high-risk PMF | Retrospective cohort study | MPN-BP: 29% ORR, 10.5 mo median OS MPN-AP: 62% ORR, 11.8 mo median OS | 41 |
| Ruxolitinib monotherapy | ||||
| Ruxolitinib | MPN-BP | Phase 2 clinical trial | ORR 16.6% | 48 |
| HMA + ruxolitinib | ||||
| Ruxolitinib + decitabine | MPN-AP, MPN-BP | Phase 2 clinical trial | ORR 44%; median OS 9.5 mo | 49 |
| MPN-BP | Phase 1/2 clinical trial | ORR 45%; median OS 8.4 mo | 50 | |
| HMA + venetoclax | ||||
| HMA + venetoclax | MPN-BP | Retrospective cohort study | Newly diagnosed: ORR 50%; median OS 7 mo R/R: no objective responses; median OS 3 mo | 45 |
| MPN-BP | Retrospective cohort study | ORR 44%; median OS 8 mo | 44 | |
| MPN-AP, MPN-BP | Retrospective multicenter cohort study | ORR: 52%; median OS 6 mo | 47 | |
| IDH1/2 inhibitors | ||||
| Enasidenib | IDH2-mutant MPN-AP, MPN-BP | Retrospective cohort study | ORR: 37.5% per ELN 2017 and 75% per MPN-BP response criteria | 54 |
| Enasidenib or ivosidenib | IDH1 or IDH2-mutant MPN-AP, MPN-BP | Retrospective cohort study | 43% CR rate among newly diagnosed patients; no objective responses in R/R cohort | 53 |
| Therapeutic regimen | Patient population | Study design | Key outcomes | Reference |
|---|---|---|---|---|
| Intensive chemotherapy | ||||
| Intensive chemotherapy (not specified) | MPN-BP | Retrospective cohort study | Median OS 10.2 mo | 61 |
| “7 + 3” cytarabine plus an anthracycline | MPN-BP | Retrospective cohort study | CR rate 67%; median OS 19.2 mo for allo-HCT vs 3.8 mo for nontransplant patients | 5 |
| AML-like induction chemotherapy | MPN-BP | Retrospective cohort study | CR rate 35%; 3-y OS 32% for allo-HCT vs 19% for nontransplant patients | 4 |
| HMA monotherapy | ||||
| Azacitidine | MPN-BP/AP | Retrospective cohort study | ORR 61.5%; median OS 13.5 mo | 43 |
| MPN-BP | Retrospective cohort study | ORR 68%; median OS 9.9 mo | 42 | |
| MPN-BP | Retrospective cohort study | Median OS 9 mo | 61 | |
| Azacitidine or decitabine | MPN-BP | Retrospective cohort study | Median OS 6.7 mo | 5 |
| Decitabine | MPN-BP, MPN-AP, high-risk PMF | Retrospective cohort study | MPN-BP: 29% ORR, 10.5 mo median OS MPN-AP: 62% ORR, 11.8 mo median OS | 41 |
| Ruxolitinib monotherapy | ||||
| Ruxolitinib | MPN-BP | Phase 2 clinical trial | ORR 16.6% | 48 |
| HMA + ruxolitinib | ||||
| Ruxolitinib + decitabine | MPN-AP, MPN-BP | Phase 2 clinical trial | ORR 44%; median OS 9.5 mo | 49 |
| MPN-BP | Phase 1/2 clinical trial | ORR 45%; median OS 8.4 mo | 50 | |
| HMA + venetoclax | ||||
| HMA + venetoclax | MPN-BP | Retrospective cohort study | Newly diagnosed: ORR 50%; median OS 7 mo R/R: no objective responses; median OS 3 mo | 45 |
| MPN-BP | Retrospective cohort study | ORR 44%; median OS 8 mo | 44 | |
| MPN-AP, MPN-BP | Retrospective multicenter cohort study | ORR: 52%; median OS 6 mo | 47 | |
| IDH1/2 inhibitors | ||||
| Enasidenib | IDH2-mutant MPN-AP, MPN-BP | Retrospective cohort study | ORR: 37.5% per ELN 2017 and 75% per MPN-BP response criteria | 54 |
| Enasidenib or ivosidenib | IDH1 or IDH2-mutant MPN-AP, MPN-BP | Retrospective cohort study | 43% CR rate among newly diagnosed patients; no objective responses in R/R cohort | 53 |
Only a minority of patients with MPN-BP are eligible for intensive chemotherapy and allo-HCT. In patients ineligible for intensive chemotherapy or allo-HCT, lower-intensity therapies that frequently use a hypomethylating agent (HMA) as monotherapy or in combination with other agents continue to be the mainstay of therapy. While the HMAs azacitidine and decitabine have been widely used in the treatment of MDS as well as AML for over a decade, the quality of evidence supporting their use in MPN-BP is limited, with no prospective, randomized data available.38-40 In small retrospective studies, the overall response rate (ORR) ranges from 29% to 61% for HMA monotherapy with median OS of 6 to 13.5 months.41-43 However, cross-study comparisons are limited by differences in patient populations and response assessments.
Inspired by the success of HMAs in combination with venetoclax in the treatment of older, intensive chemotherapy- ineligible patients with AML, this combination has been used off-label in patients with MPN-BP as well. In a retrospective, multicenter study of 32 MPN-BP patients treated with HMAs plus venetoclax, the rate of complete remission (CR) with incomplete count recovery (CRi) was 44% for the combination compared to 4% with an HMA alone in a historic control group. However, the median OS was 8 months for patients treated with an HMA plus venetoclax and was not statistically significantly better than HMA alone.44 In a similar study from MD Anderson Cancer Center, the CR rate with an HMA plus venetoclax was 43% among 14 patients with newly diagnosed post-MPN AML, resulting in a median OS of 7 months.45 Similar to studies in relapsed/refractory (R/R) AML, an HMA plus venetoclax had only limited efficacy in patients with R/R MPN-BP, with no objective responses seen and a median OS of 3 months.45,46 In another multicenter, retrospective analysis of both treatment-naive and R/R MPN-BP patients (n = 27) treated with venetoclax-based therapies, the ORR was 53%.47 However, this high response rate did not lead to a durable survival benefit, with a median OS of 6 months and an inferior survival rate (HR, 3.42; 95% CI, 1–11.8; P = .03) in patients with TP53 mutations.47 While HMA plus venetoclax combinations can be an option for some patients with MPN-BP, especially as a bridge to allo-HCT, additional studies are needed to inform patient selection based on molecular disease characteristics as well as potential combination therapies (eg, NCT03862157).
While the Jak inhibitor ruxolitinib has led to significant improvements in the treatment of patients with chronic-phase myelofibrosis, the role of ruxolitinib in MPN-BP is less clear, with limited efficacy if used as a single agent in MPN-BP.7,48 In a recent phase 2 study of ruxolitinib in combination with decitabine (NCT02076191), 25 patients with MPN-BP/MPN-AP received ruxolitinib (25 mg twice a day during induction followed by 10 mg twice a day during subsequent cycles) in combination with decitabine administered at 20 mg/m2 for 5 consecutive days on a 28-day cycle. The ORR, defined as a composite of CR, CRi, and partial remission, was 44% (11 out of 25 patients), with a median OS of 9.5 months (95% CI, 4.3-12.0 months).49 Treatment was generally well tolerated, with febrile neutropenia (7 patients, 28%), pneumonia (6 patients, 24%), and anemia (4 patients, 16%) being the most common grade 3/4 adverse events.49 These results were comparable to another single-center phase 2 study reporting an ORR of 45% with a median OS of 8.4 months.50
Future directions
Advances in the understanding of the underlying pathophysiology and molecular landscape have enabled the identification of novel therapeutic targets that are currently under investigation in clinical trials. Table 3 summarizes ongoing clinical trials in MPN-BP. Among the novel targets, BET inhibitors and MDM2 inhibitors have shown preclinical activity in MPN-BP models, but no clinical trial data are available to date.31,51,52 Only a small subset of MPN-BP patients with targetable mutations in IDH1 or IDH2 may benefit from treatment with the IDH1 inhibitor ivosidenib or the IDH2 inhibitor enasidenib, either as monotherapy or in combination with other agents. In a series of 12 patients treated with IDH1/2 inhibitor–based combinations, 3 out of 7 patients with newly diagnosed MPN-BP achieved a CR, while none of the patients with R/R disease responded.53 Another retrospective study of 8 patients with IDH2-mutant MPN-BP treated with enasidenib reported an ORR of 37.5% per European LeukemiaNet (ELN) 2017 and 75% per MPN-BP response criteria.54 Besides the need for additional prospective studies to further define the safety and efficacy of IDH inhibitors in MPN-BP and to identify potentially synergistic combination therapies, this latter study highlights an important aspect of clinical trial design and interpretation in MPN-BP. Current ELN 2017 response criteria for AML mandate the absence of blasts from the peripheral blood for a CR.55 However, a small percentage of circulating blasts are frequently seen in patients with chronic-phase MPNs, and it might be unrealistic to expect patients with MPN-BP to achieve peripheral blood blast clearance because treatment may not reverse the underlying fibrosis in the bone marrow and the extramedullary hematopoiesis underlying this small peripheral blast population. Converting patients back to a chronic-phase MPN state with hematologic improvement may provide important benefits to patients independent of the more stringent AML response criteria, which has led to the development of dedicated response criteria for MPN-BP. However, uptake in both routine clinical practice and trials has been limited.56
Overview of selected ongoing clinical trials in MPN-BP
| Agent | NCT | Phase | Patient population |
|---|---|---|---|
| Decitabine + ruxolitinib or fedratinib | NCT04282187 | 2 | MPN-AP and MPN-BP as bridge to allo-HCT |
| Ruxolitinib + enasidenib | NCT04281498 | 2 | IDH2-mutant MPN-AP, MPN-BP, chronic-phase myelofibrosis |
| Fedratinib + ivosidenib or enasidenib | NCT04955938 | 1 | IDH1- or IDH2-mutant MPN with ≥5% blasts |
| Azacitidine + venetoclax | NCT05074355 | 2 | MPN-AP and MPN-BP |
| KRT-232 (MDM2 inhibitor) | NCT04113616 | 1/2 | post-MPN AML |
| ZN-d5 (BCL2 inhibitor) + ZN-c3 (WEE1 inhibitor) | NA | 1/2 | AML including post-MPN AML |
| Agent | NCT | Phase | Patient population |
|---|---|---|---|
| Decitabine + ruxolitinib or fedratinib | NCT04282187 | 2 | MPN-AP and MPN-BP as bridge to allo-HCT |
| Ruxolitinib + enasidenib | NCT04281498 | 2 | IDH2-mutant MPN-AP, MPN-BP, chronic-phase myelofibrosis |
| Fedratinib + ivosidenib or enasidenib | NCT04955938 | 1 | IDH1- or IDH2-mutant MPN with ≥5% blasts |
| Azacitidine + venetoclax | NCT05074355 | 2 | MPN-AP and MPN-BP |
| KRT-232 (MDM2 inhibitor) | NCT04113616 | 1/2 | post-MPN AML |
| ZN-d5 (BCL2 inhibitor) + ZN-c3 (WEE1 inhibitor) | NA | 1/2 | AML including post-MPN AML |
NA, not available.
CLINICAL CASE (Continued)
Repeat molecular testing at the time of progression to MPN-BP showed IDH2, NPM1, TET2, and SRSF2. Following initial cytoreduction with hydroxyurea, the patient started treatment with azacitidine and venetoclax. Treatment with azacitidine and venetoclax was complicated by neutropenic sepsis and prolonged neutropenia and thrombocytopenia requiring hospital admission and transfusion support. Following 2 cycles of azacitidine and venetoclax, the patient achieved a complete acute leukemia response per the definitions proposed by the Post-Myeloproliferative Neoplasm Acute Myeloid Leukemia Consortium.56 He continues on azacitidine and venetoclax and is undergoing evaluation for an allo-HCT.
Conclusion
Patients with chronic-phase MPNs have a variable risk of progression to MPN-AP or MPN-BP, defined as an increase in blast count of 10% to 19% and greater than or equal to 20%, respectively. Adverse cytogenetic and molecular features are enriched in patients with MPN-BP, contributing to poor outcomes. While allo-HCT remains the only potentially curative therapeutic modality for patients with MPN-BP, only a minority of patients are eligible for this intense treatment modality. HMAs alone or in combination with Jak inhibitors or venetoclax, as well as IDH1/2 inhibitors in IDH1/2-mutant patients, can yield objective responses in a subset of patients. Several novel agents are in various stages of clinical development but are not yet available for routine use at this point, highlighting the need for ongoing research and the prioritization of clinical trial enrollment when feasible.
Conflict-of-interest disclosure
Jan Philipp Bewersdorf: no competing financial interests to declare.
Raajit K. Rampal: research funding: Constellation, Incyte, Stemline, Zentalis; consultancy: Abbvie, Blueprint, Celgene/Bristol Myers Squibb, Constellation, CTI, Disc Medicines, Galecto, Incyte, Jazz, Novartis, Pharmaessentia, Promedior, Sierra Oncology, Stemline, Sumitomo Pharma, Zentalis.
Off-label drug use
Jan Philipp Bewersdorf: nothing to disclose.
Raajit K. Rampal: nothing to disclose.
