

Abstract
Patients with chronic lymphocytic leukemia (CLL) refractory to covalent BTK and BCL2 inhibitors have a new unmet clinical need. Standard treatment options are able to obtain only limited and short-lasting disease control associated with reduced overall survival, and thus these patients have become ideal candidates for enrollment in clinical trials. Favorable results have been obtained with the use of noncovalent BTK inhibitors (roughly 70% overall response rate regardless of the actual resistance or intolerance to previous covalent BTK inhibitors) and anti-CD19 chimeric antigen receptor (CAR) T-cell therapy (with complete responses in up to 45% of cases and an undetectable measurable residual disease rate of 65% in the bone marrow). These 2 approaches should be considered valid options in this setting, although not yet approved. For young fit patients achieving remissions with salvage treatments, the option of allogeneic stem cell transplantation should be discussed as the outcome appears to be unaffected by number and type of previous targeted agents. Novel treatment strategies interfering with different mechanisms of CLL cell survival and proliferation are warranted, including small molecules with novel targets (eg, CDK9, MCL1, ERK inhibitors), CAR T cells targeting different antigens, CAR natural killer cells, or bispecific antibodies.
Learning Objectives
Patients relapsing after covalent BTK inhibitors and venetoclax are a new unmet clinical need with a dismal outcome and reduced overall survival
More robust data are currently available for noncovalent BTK inhibitor and anti-CD19 chimeric antigen receptor T-cell therapy; novel promising targets are on the horizon
CLINICAL CASE
Ms Green was diagnosed with chronic lymphocytic leukemia (CLL) in 2005, at only 44 years of age. Her disease was staged as Rai I/Binet A, and a watch-and-wait policy was advised. Two years later, she experienced disease progression with increased lymph nodes; was treated with fludarabine, cyclophosphamide, and rituximab (FCR) for 6 cycles; and achieved a complete remission (CR). Prior to the initiation of treatment, biomarker assessment revealed that the patient harbored an unmutated immunoglobulin heavy-variable gene (IGHV); no del(13q), del(11q), del(17p), and trisomy 12; and no mutations within the TP53 gene. Five and a half years after completing FCR, she relapsed with anemia and thrombocytopenia due to bone marrow (BM) infiltration. Molecular and cytogenetic testing was repeated and detected no aberrations by fluorescence in situ hybridization and no mutations within the TP53 gene. She received 6 cycles of bendamustine with rituximab (BR) and NOXA12, a CXCL12 inhibitor within a clinical trial, and obtained a partial response (PR).1 Nine months after completing BR + NOXA12, she relapsed with hyporegenerative anemia and thrombocytopenia. She commenced treatment with idelalisib + rituximab within the GS-US-312-1325 study.2 Idelalisib was discontinued ~2 years after treatment initiation due to gastrointestinal toxicity and having achieved a PR. A few months after discontinuation of idelalisib, she again experienced disease progression and started treatment with ibrutinib that was continued for 24 months, without relevant treatment-related toxicities and achieving a PR. At that time, anemia and thrombocytopenia co-occurred with progressively increasing abdominal lymph nodes. She was switched to venetoclax, the ramp-up of which she completed with great challenges due to rapid disease flare upon ibrutinib discontinuation. TP53 mutation was tested again prior to the start of treatment with venetoclax, and 2 different TP53 mutations (within exons 7 and 11) were identified together with trisomy at chromosome 17, incorporating the TP53 locus, in 25% of cells. The BTK gene was not sequenced, and hence no information on BTK resistance mutations was available. Venetoclax was very well tolerated and led to a PR (small residual abdominal lymph nodes up to 2 cm persisted). Two and a half years after starting full-dose venetoclax, her disease progressed again due to anemia and thrombocytopenia, as well as enlarged abdominal lymph nodes. In October 2021, 16 years postdiagnosis, she is now 60 years old, has no other relevant comorbidities, but has failed chemoimmunotherapy (FCR and BR + NOXA12), PI3K inhibitors (idelalisib + rituximab) due to intolerance, a covalent BTK inhibitor (cBTK inhibitor, ibrutinib) and a BCL2 inhibitor (venetoclax), both due to progression under treatment. How would you manage the patient at this point?
BTK inhibitor and BCL2 inhibitor refractory patients with CLL: recognizing a new unmet clinical need
The management of patients with CLL has radically changed over the past 15 years following the introduction of novel targeted agents. Owing to the approval and wide availability of covalent BTK inhibitors (BTKi; ie, ibrutinib, acalabrutinib, and [zanubrutinib, although not yet approved for use in patients with CLL]), PI3K inhibitors (eg, idelalisib or duvelisib), and BCL2 inhibitors (BCL2i) such as venetoclax, opportunities for long-term disease control with tolerable and effective targeted treatments exist.3 This has led to a landmark shift in the clinical management of CLL from the therapeutic paradigm of the past decades, which was based on chemoimmunotherapy (CIT) (namely, FCR, BR, chlorambucil ± anti-CD20 monoclonal antibody).4
The patient's therapeutic trajectory described in the Clinical Case detailed above (first- and second-line CIT followed by the sequencing of targeted agents) is unlikely to be encountered in clinical practice over the coming years because we now have approved targeted agents in both the frontline and the relapsed/refractory setting. That notwithstanding, data published on novel targeted agents predominantly derive from patients who were previously exposed to CIT before receiving targeted inhibitor, with <1% of patients (9/921) included in 6 landmark clinical trials using covalent BTKi or BCL2i in relapsed/refractory CLL previously exposed to targeted agents.5
The 2 main reasons for discontinuing targeted agents are (1) resistance or (2) intolerance, with intolerance evidenced more frequently in the real-world setting compared to clinical trials.
Discontinuation of treatment due to intolerance in the real world is reported in up to 26% to 41% of patients treated with ibrutinib6,7 and 6% to 29% of patients treated with venetoclax- based combinations.8,9 Importantly, if the patient discontinues treatment with kinase inhibitors for intolerance (as detailed in our Clinical Case; ie, idelalisib discontinuation), there is a window of opportunity for the administration of alternative kinase inhibitors. As mentioned above, our patient was subsequently started on ibrutinib, but the same approach can be applied in patients discontinuing ibrutinib due to intolerance, as second-generation covalent BTKi, such as acalabrutinib and zanubrutinib, appear to be well tolerated and only infrequently lead to the same toxicity experienced with the first-generation inhibitor.10–13
BTKi resistance is definitely a challenging scenario, and while molecular mechanisms of resistance have been identified in some patients, we still lack knowledge regarding resistance in a substantial proportion.14 In up to 80% of cases, resistance to ibrutinib (with preliminary evidence also presented for acalabrutinib15 and zanubrutinib16 ) is associated with the detection of mutations within the BTK and/or PLCG2 genes. BTK mutations (most frequently involving the key residue cysteine 481, C481) disrupt the covalent binding of the drug, while PLCG2 mutations are mainly gain-of-function mutations (PLCG2 lies downstream to BTK). These mutations lead to B-cell receptor signaling activation even in the presence of therapeutic levels of the covalent BTKi.
Resistance to venetoclax is more elusive with marked intra- and interpatient heterogeneity. BCL2 mutations have been detected in 25% to 50% of cases, together with overexpression of alternative prosurvival proteins (MCL1, BCL-XL) and metabolic reprogramming.17,18 More recently, activation of the nuclear factor–κB pathway has been suggested as a driver of resistance in patients receiving continuous venetoclax19 as well as dysregulation and epigenetic modification of PUMA.20
A comprehensive, although disappointing and anecdotal, list of treatment options in patients who have failed both BTKi and BCL2i is currently available in standard clinical practice and includes PI3Ki, cytotoxic chemotherapy, alemtuzumab, high-dose methylprednisolone with or without anti-CD20 antibodies, single-agent anti-CD20 antibodies, and allogeneic stem cell transplantation (allo-SCT) (this last option is restricted to younger, highly selected patients based on their comorbidities and performance status).
The PI3Ki idelalisib and duvelisib were approved based on studies that solely comprised patients treated with CIT, and thus their efficacy in the setting of patients previously treated with BTKi and BCL2i may be limited. In 17 patients treated with PI3Ki after prior sequential covalent BTKi and BCL2i therapies, the overall response rate (ORR) was 46.9% with a median progression-free survival (PFS) of 5 months; 78% of patients discontinued treatment, mainly due to disease progression.8 The use of PI3Ki is also limited by the significant toxicity observed, in terms of immune-mediated colitis, pneumonitis, and infections, resulting in a high adverse event (AE)–related discontinuation rate (in the ASCEND study over a 4-year follow-up period, 67% of patients had to discontinue treatment with idelalisib due to AEs).21
No data on the use of CIT in patients relapsing after BTKi and BCL2i are available, although anecdotal experience suggests that disease control is inadequate and virtually nonexistent if the disease has evolved and acquired poor-prognostic genetic events such as TP53 aberrations.
While retreatment with a BTKi after venetoclax discontinuation seems a reasonable option in patients who interrupted the first covalent BTKi due to intolerance (ORR at retreatment 70% in BTKi-intolerant patients) and translates into prolonged disease control (median PFS not reached), it achieves less and only short-lasting responses (ORR, 50%; median PFS, 4 months) in BTKi-refractory patients,8 including, as expected, heavily pretreated individuals with high-risk disease.
What, then, are our options for the management of BTKi- and BCL2i-refractory patients?
Noncovalent BTKi
The future is not dismal at all, and over the past few years, novel agents and treatment strategies have been devised. Noncovalent reversible BTKi has been tested recently in clinical trials and demonstrated favorable outcomes in patients who have exhausted all currently available treatment options (ie, refractory to CIT, covalent BTKi, BCL2i, and PI3Ki) (Table 1). The preliminary efficacy data of noncovalent BTKi provide a proof of principle that, in many instances, CLL maintains dependence on B-cell receptor signaling mediated by BTK after progression on covalent BTKi, lending support to the development of additional agents that would act on this pathway. The compounds leading the way in this arena are pirtobrutinib and nemtabrutinib.
Noncovalent BTKi currently in clinical trials
| Name | Number of patients with R/R CLL in pivotal trials | ORR, % | Median PFS | Median follow-up |
|---|---|---|---|---|
| Pirtobrutinib22,23 | 252 (evaluable for response) | 68 | NR | 9 mo |
| Nemtabrutinib24 | 38 (evaluable for response) | 57.9 | NE | 4.6 mo |
| Vecabrutinib* 46 | 30 | 3 | NE | 28.5 wk† |
| Name | Number of patients with R/R CLL in pivotal trials | ORR, % | Median PFS | Median follow-up |
|---|---|---|---|---|
| Pirtobrutinib22,23 | 252 (evaluable for response) | 68 | NR | 9 mo |
| Nemtabrutinib24 | 38 (evaluable for response) | 57.9 | NE | 4.6 mo |
| Vecabrutinib* 46 | 30 | 3 | NE | 28.5 wk† |
Terminated.
Median time on treatment for patients with partial response/stable disease.
NE, not estimable; NR, not reached; R/R, relapsed/refractory.
Pirtobrutinib was developed as an orally available, highly selective, reversible BTKi with an equally low nM potency against wild-type and C481-mutant BTK in preclinical studies. Updated results of the phase 1/2 BRUIN study showed that in 252 heavily pretreated patients with CLL (86% previously exposed to BTKi, 75% of whom discontinued BTKi treatment due to progression), the ORR was 68%.22,23 Response to pirtobrutinib did not differ between patients previously exposed to both BTKi and BCL2i and was as high as 71% in patients carrying a hotspot BTK mutation (p.C481S). The drug was effective also in patients carrying PLCG2 mutations, thus suggesting that the relevance of this aberration in the context of BTKi resistance still needs to be elucidated further. Of particular note, pirtobrutinib-related AEs were few, mainly grade 1 to 2, and included fatigue (20%), diarrhea (17%), and contusion (13%), with no evidence of increased cardiovascular toxicity. Similar, albeit preliminary, data were recently presented for nemtabrutinib (MK-1026, aka ARQ-531), a noncovalent BTKi24 that showed that a population of heavily pretreated relapsed/refractory patients with CLL (median number of prior therapies, 4; 84% previously treated with a covalent BTKi; 63% carrying the hotspot C481S BTK mutation) obtained an ORR of 58% after a median follow-up of 4.56 months. Nemtabrutinib-associated toxicity, although less mild compared to pirtobrutinib, was generally manageable, with dysgeusia (15%), nausea (11%), fatigue (11%), and decreased neutrophil count (10%) being the most common drug-related AEs.
Chimeric antigen receptor T-cell therapy
The use of chimeric antigen receptor (CAR) T-cell therapy is also emerging as a promising treatment strategy for patients with relapsed/refractory disease, particularly those treated with BTKi/BCL2i.
Initial results with CAR T cells in patients with CLL were impressive,25 and more than a decade after the administration of anti-CD19 CAR T cells in 3 patients with CLL, 2 of the patients achieved a CR and remained progression free with the persistence of anti-CD19 CAR T cells.26 Although not exposed to BTKi or BCL2i, these 2 patients had received numerous treatment lines and, at the time of the administration of CAR T cells, had exhausted all alternative treatment options, thus supporting the potential of CAR T cells in the setting of patients relapsing/refractory to BTKi and BCL2i. That notwithstanding, after the initial successful experience, older age, comorbidity burden, and paradigmatic CLL-related T-cell dysfunction have emerged as shortcomings hampering CAR T-cell development and its wide application in CLL. In order to overcome these hurdles, several strategies have been pursued with positive initial results (Table 2).27–31 In a recent retrospective series, 18 patients previously exposed to covalent BTKi and venetoclax received CAR T-cell therapy and obtained an ORR of 66%, with a median PFS of 9 months.8 In the phase 1 monotherapy dose-escalation portion of the TRANSCEND CLL 004 study, lisocabtagene maraleucel (liso-cel), an autologous CD19-directed CAR T-cell product that has a defined composition of equal target doses of CD8+ and CD4+ T cells, was administered to 23 patients with relapsed/refractory CLL.27 Patients with high-risk features, including TP53 aberrations, unmutated IGHV, and/or complex karyotype, had been previously exposed to ≥2 lines of therapy while standard-risk patients had been treated with ≥3 lines. All patients had undergone prior treatment with ibrutinib, and 65% of them were previously treated with both ibrutinib and venetoclax. The ORR in 22 evaluable patients was 82%, with a CR/CR with incomplete bone marrow recovery (CRi) rate of 45%, and undetectable measurable residual disease (uMRD) rate in peripheral blood (PB) by flow was 75%. Of note, 65% of patients also achieved uMRD in the BM when analyzed by next-generation sequencing, and those patients exhibiting uMRD, in both the PB and BM, and achieving a PR/CR according to standard iwCLL2018 criteria had the longest PFS (not reached vs 3 months in those patients demonstrating measurable residual disease positivity), thus indicating that achieving uMRD might be a valuable goal in this setting and may predict long-term efficacy of CAR T cells. This result is even more impressive considering the difficult-to-treat patient cohort included in the study, with 83% harboring high-risk features, 91% being refractory to ibrutinib, and 48% having failed both ibrutinib and venetoclax. Following treatment, 23% (5/22) later progressed to Richter transformation, with a median PFS of 18 months. Analysis of the subgroup of 10 evaluable patients who progressed on BTKi and failed venetoclax showed an ORR of 80%, with 60% CR/CRi and 78% and 67% uMRD in the PB and BM, respectively, with a median duration of response of 17 months and a median PFS of 13 months. This study also included 2 additional cohorts, one based on the combination of liso-cel with ibrutinib and a second one combining liso-cel with venetoclax. Preliminary results of the cohort receiving liso-cel and ibrutinib reported an even higher ORR of 95%, with a 47% CR/CRi rate.31
Key recent studies exploring CAR T-cell therapy in CLL
| CAR T-cell therapy | Number of patients with CLL | Prior cBTKi/prior BCL2i | ORR/uMRD in BM, % | Grade ≥3 CRS/grade ≥3 ICANS, % |
|---|---|---|---|---|
| CD19-4-1BB27 | 24 | 19/6 | 71/58* | 8/25 |
| CD19-4-1BB24 | 14 (infused) | 1/NA | 57/21* | 43/7 |
| CD19-4-1BB with ibrutinib29 | 19 | 19/11 | 83/72† | 0/26 |
| CD19-4-1BB26 | 23 | 23/15 | 82/65* | 9/17 |
| CAR T-cell therapy | Number of patients with CLL | Prior cBTKi/prior BCL2i | ORR/uMRD in BM, % | Grade ≥3 CRS/grade ≥3 ICANS, % |
|---|---|---|---|---|
| CD19-4-1BB27 | 24 | 19/6 | 71/58* | 8/25 |
| CD19-4-1BB24 | 14 (infused) | 1/NA | 57/21* | 43/7 |
| CD19-4-1BB with ibrutinib29 | 19 | 19/11 | 83/72† | 0/26 |
| CD19-4-1BB26 | 23 | 23/15 | 82/65* | 9/17 |
By IGH sequencing.
By flow cytometry.
CRS, cytokine release syndrome; ICANS, immune effector cell-associated neurotoxicity syndrome; NA, not available.
Allogeneic stem cell transplant
Data on allo-SCT in the double-refractory setting are very scant as the indication for allo-SCT was mainly related to the presence of TP53 aberrations.
More recently, 2 different studies focused on the outcome of patients who received allo-SCT having previously been treated with ≥1 targeted agent(s), thus being frequently double-exposed to cBTKi and BCL2i but only seldom double-refractory, considering the need of a bridging therapy before allo-SCT in order to obtain disease control. Of the 108 patients included in one of these series, 30 were previously exposed to targeted agents and demonstrated a very favorable outcome upon transplant with a 3-year PFS and 3-year overall survival (OS) of 69% and 87%, respectively. The procedure was very well tolerated with a 3-year cumulative incidence of nonrelapse mortality of only 7% and a cumulative incidence of relapse of 24%.32 Similarly, in a series of 65 patients who were previously exposed to targeted agents (13% double-refractory) and subsequently underwent allo-SCT,33 24-month PFS and 24-month OS were estimated to be 63% and 81%, respectively. In line with the eligibility criteria for allo-SCT, these patients were younger than the general CLL patient population and had a high prevalence of unfavorable disease features (51% carried TP53 mutations, 44% had del(17p), 90% expressed unmutated IGHV, and 50% had complex karyotypes). Of note, 29% of patients in this series had exclusively received targeted agents (82% ibrutinib, 40% venetoclax, and 20% idelalisib). At least in part due to the reduced intensity conditioning regimen administered in 95% of cases, nonrelapse mortality at 24 months was as low as 13%. Long-term toxicity (ie, moderate to severe chronic graft-vs-host disease) occurred in 27% of patients; on the other hand, about 27% of patients experienced disease relapse over time. Notably, no differences in PFS and OS were observed in patients who had received ibrutinib compared to venetoclax as a bridge to allo-SCT. Multivariate analyses confirmed that only a higher hematopoietic cell transplantation–specific comorbidity index was predictive for shorter PFS.
Novel therapies and combinations on the horizon
Novel potential treatment strategies and combinations are currently being explored to improve the outcome of cBTKi- and BCL2i-exposed patients with CLL, including the following (Table 3 and Figure 1):
Bispecific antibody combining anti-CD3 and anti-CD20 (mosunetuzumab, epcorizumab), inducing potent activation and cytotoxic activity of CD4+ and CD8+ T cells to selectively eliminate CD20-expressing cells37
Targeting BTK degradation by chimeric degradation activation compound active against both wild-type and mutant BTK38
CAR T cells and bispecific antibodies against novel targets (eg, ROR1, a transmembrane protein in the receptor tyrosine kinase–like orphan receptor—ROR—family, expressed at high levels on most CLL cells and absent from most normal B cells)
Human leukocyte antigen–mismatched anti-CD19 CAR natural killer cells,39 which have been reported to induce a response in 4 of 5 patients with CLL (1 with concomitant Richter transformation) with 3 CR; all responding patients remained alive at a median follow-up of 13.8 months
Small molecules targeting novel key mechanisms (eg, CDK9, MCL1, ERK)
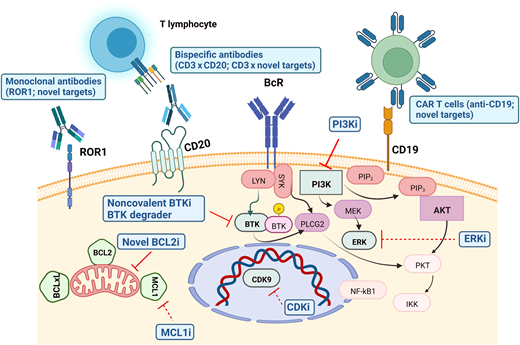
Promising therapeutic targets in BTKi- and BCL2i-resistant patients with CLL. Mechanisms of inhibition in advanced clinical development are depicted with solid lines, while those with more preliminary data are represented in dotted lines. CDKi, cyclin-dependent kinase inhibitors; ERKi, extracellular signal–regulated kinase inhibitors; MCL1i, myeloid leukemia 1 inhibitors. The figure was created with BioRender.
Promising therapeutic targets in BTKi- and BCL2i-resistant patients with CLL. Mechanisms of inhibition in advanced clinical development are depicted with solid lines, while those with more preliminary data are represented in dotted lines. CDKi, cyclin-dependent kinase inhibitors; ERKi, extracellular signal–regulated kinase inhibitors; MCL1i, myeloid leukemia 1 inhibitors. The figure was created with BioRender.
Experimental agents under development for patients with relapsed/refractory CLL
| Name | Mechanism | ORR in patients with R/R CLL | Currently evaluated in BKTi/BCL2i refractory patients with CLL | Clinical trial registration number |
|---|---|---|---|---|
| Lisaftoclax | BCL2 inhibitor | Not available | No | NCT05147467 |
| BGB-11417 | BCL2 inhibitor | 2/6 (monotherapy) 12/19 (in combination with zanubrutinib) | Yes | NCT04277637 |
| LOXO-338 | BCL2 inhibitor | Not available | No | NCT05024045 |
| Epcoritamab | CD20 × CD3 bispecific monoclonal antibody | Not available | Yes | NCT04623541 |
| Mosunetuzumab | CD20 × CD3 bispecific monoclonal antibody | Not available | Yes | NCT05091424 |
| BGB-16673 | BTK degrader | Not available | Yes | NCT05006716 |
| NX-2127 | BTK degrader and IMiD | Not available | Yes | NCT04830137 |
| NX-5948 | BTK degrader | Not available | Yes | NCT05131022 |
| Zilovertamab | Anti-ROR1 monoclonal antibody | 31/34 (including both 12 TN and 22 R/R patients) | No | NCT03088878 |
| VIP152 | CDK9 inhibitor | Not available | Yes | NCT04978779 |
| Name | Mechanism | ORR in patients with R/R CLL | Currently evaluated in BKTi/BCL2i refractory patients with CLL | Clinical trial registration number |
|---|---|---|---|---|
| Lisaftoclax | BCL2 inhibitor | Not available | No | NCT05147467 |
| BGB-11417 | BCL2 inhibitor | 2/6 (monotherapy) 12/19 (in combination with zanubrutinib) | Yes | NCT04277637 |
| LOXO-338 | BCL2 inhibitor | Not available | No | NCT05024045 |
| Epcoritamab | CD20 × CD3 bispecific monoclonal antibody | Not available | Yes | NCT04623541 |
| Mosunetuzumab | CD20 × CD3 bispecific monoclonal antibody | Not available | Yes | NCT05091424 |
| BGB-16673 | BTK degrader | Not available | Yes | NCT05006716 |
| NX-2127 | BTK degrader and IMiD | Not available | Yes | NCT04830137 |
| NX-5948 | BTK degrader | Not available | Yes | NCT05131022 |
| Zilovertamab | Anti-ROR1 monoclonal antibody | 31/34 (including both 12 TN and 22 R/R patients) | No | NCT03088878 |
| VIP152 | CDK9 inhibitor | Not available | Yes | NCT04978779 |
CDK9, cyclin-dependent kinase 9; IMiD, immunomodulatory drug; TN, treatment-naive.
Recently, 2 retrospective series reported the use of combined ibrutinib and venetoclax in cBTKi/BCL2i-refractory patients.40,41 In 1 series, all 11 double-refractory patients responded to this combination (clinical CR, 55%; PR, 45%); although median duration of treatment was only 7.5 months, and median time to next treatment 11.2 months, the combination of ibrutinib and venetoclax led to a median OS of 27 months.40 In the second cohort, 4 of 6 patients deemed truly refractory to BTKi and venetoclax single agents were treated with combined BTKi and venetoclax and achieved a PR lasting between 9.3 and 44.2 months.41 In this setting, even transient responses are meaningful as a bridge to cellular therapy or novel clinical trials.
CLINICAL CASE (Continued)
Ms Green was enrolled on the NCT03740529 phase 1/2 study in October 2021. She was started on pirtobrutinib 200 mg once a day with rapid reduction of lymph nodes and an improvement in her anemia and platelet values. In May 2022, 8 months after treatment initiation, her complete blood cell count was normal (white blood cell count, 5.8 × 109/L; absolute neutrophil count, 2.2 × 109/L; absolute lymphocyte count, 3.1 × 109/L; hemoglobin, 13.8 g/dL; platelets, 168 × 109/L), and only small (1 cm) superficial lymph nodes were detected by clinical examination; response assessment according to iwCLL2018 criteria showed a PR.
Conclusion
Recent evidence suggests that salvage therapy after failing covalent BTKi and BCL2i remains suboptimal.42 Noncovalent BTKi and cellular therapies appear to be the most effective step forward, but they are currently only available through clinical trials. Further studies need to be conducted in order to identify novel potential targets (eg, ROR1) and novel mechanisms of action (eg, bispecific antibodies) that may regain long-term disease control in this group of patients. Although our current knowledge mainly stems from patients previously exposed to CIT and receiving targeted agents as continuous treatments, what we can envisage in the future is that combination strategies of targeted agents will allow safe treatment “holidays” and retreatment with the same agent(s) upon disease relapse, therefore preventing the occurrence of resistant disease due to continuous clonal pressure. This scenario has, to date, only been tested in patients relapsing after first-line fixed-duration treatment with ibrutinib and venetoclax (CAPTIVATE study)43 or venetoclax + obinutuzumab44 and in the relapsed/refractory setting, after failing a venetoclax and rituximab 24-month regimen in the MURANO study.45
While much work needs to be done, new agents hold great promise for this currently difficult-to-treat group of patients.
Acknowledgments
The author thanks Prof. Paolo Ghia for his constant support, valuable insights, and mindful revision of the manuscript; Dr. Cristina Scielzo for assisting in the design of Figure 1; and Dr. Lesley Ann Sutton for her helpful revision of the manuscript.
Conflict-of-interest disclosure
Lydia Scarfò received honoraria for advisory board participation from AbbVie, AstraZeneca, BeiGene, and Janssen and travel grants from Beigene and Janssen; she is on the speakers bureau for Octapharma.
Off-label drug use
Lydia Scarfò: pirtobrutinib, nemtabrutinib, liso-cel, and other experimental agents listed in Table 3 and in the section “Novel therapies and combinations on the horizon” are discussed, and none of these drugs are yet FDA-approved at the time of writing.
