

Abstract
Myelodysplastic syndromes (MDS) are myeloid neoplasms characterized by morphologic dysplasia, persistent cytopenia, and a variable risk of evolution to acute myeloid leukemia (AML). Risk stratification is crucial in a patient-centered approach to the treatment of MDS. Based on hematologic parameters and cytogenetic abnormalities, the Revised International Prognostic Scoring System is currently used for this purpose. In the past years, the use of massively parallel DNA sequencing has clarified the genetic basis of MDS and has enabled development of novel diagnostic and prognostic approaches. When conventional cytogenetics is combined with gene sequencing, more than 90% of patients are found to carry a somatic genetic lesion. In addition, a portion of patients has germline variants that predispose them to myeloid neoplasms. The recently developed International Consensus Classification of MDS includes new entities that are molecularly defined—namely, SF3B1-mutant and TP53-mutant MDS. The International Working Group for Prognosis in MDS has just developed the International Prognostic Scoring System–Molecular (IPSS-M) for MDS, which considers hematologic parameters, cytogenetic abnormalities, and somatic gene mutations. The IPSS-M score is personalized and can be obtained using a web-based calculator that returns not only the individual score but also the expected leukemia-free survival, overall survival, and risk of AML transformation. Providing an efficient risk stratification of patients with MDS, the IPSS-M represents a valuable tool for individual risk assessment and treatment decisions.
Learning Objectives
Understand how genomic profiling may improve clinical decision making in MDS
Prepare for the use of the International Prognostic Scoring System–Molecular in clinical practice and clinical trials
Learn how to develop precision treatments in myelodysplastic syndromes
Introduction
Myelodysplastic syndromes (MDS) are myeloid neoplasms characterized by morphologic dysplasia in 1 or more bone marrow cell lineages, persistent cytopenia in 1 or more peripheral blood cell lineages, and a variable risk of evolution to acute myeloid leukemia (AML).1 Based on the number of dysplastic lineages, presence of ring sideroblasts, percentage of bone marrow and peripheral blood blasts, and type of cytogenetic abnormality, the 2016 revision of the World Health Organization (WHO) categorizes MDS into distinct subtypes, as illustrated in Table 1.2
Classifications of MDS and premalignant cytopenias
| Classification | MDS subtypes or related conditions |
|---|---|
| 2016 WHO classification (revised fourth edition)2 | • MDS with single-lineage dysplasia (MDS-SLD) • MDS with multilineage dysplasia (MDS-MLD) • MDS with ring sideroblasts, with single lineage dysplasia (MDS-RS-SLD) or multilineage dysplasia (MDS-RS-MLD) • MDS with isolated del(5q) (MDS del (5q)) • MDS with excess blasts (MDS-EB), type 1 (MDS-EB-1) or type 2 (MDS-EB-2) • MDS, unclassifiable (MDS-U) |
| International Consensus Classification (2022)17 | MDS subtypes • MDS with mutated SF3B1 (MDS-SF3B1) • MDS with del(5q) (MDS del(5q)) • MDS, NOS—without dysplasia • MDS, NOS—with single-lineage dysplasia • MDS, NOS—with multilineage dysplasia • MDS with excess blasts (MDS-EB) • MDS with mutated TP53 • MDS/AML |
| Related clonal disorders • Clonal hematopoiesis of indeterminate potential (CHIP) • Clonal cytopenia of undetermined significance (CCUS) • VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome: autoinflammatory syndrome associated with anemia and clonal hematopoiesis due to somatic mutation in the UBA1 gene | |
| 2022 WHO classification (fifth edition)20 | MDS with defining genetic abnormalities • MDS with low blasts and isolated 5q deletion (MDS-5q) • MDS with low blasts and SF3B1 mutation (MDS-SF3B1) • MDS with biallelic TP53 inactivation (MDS-bi TP53) MDS, morphologically defined • MDS with low blasts (MDS-LB) • MDS, hypoplastic (MDS-h) • MDS with increased blasts (MDS-IB, including MDS with fibrosis) |
| Classification | MDS subtypes or related conditions |
|---|---|
| 2016 WHO classification (revised fourth edition)2 | • MDS with single-lineage dysplasia (MDS-SLD) • MDS with multilineage dysplasia (MDS-MLD) • MDS with ring sideroblasts, with single lineage dysplasia (MDS-RS-SLD) or multilineage dysplasia (MDS-RS-MLD) • MDS with isolated del(5q) (MDS del (5q)) • MDS with excess blasts (MDS-EB), type 1 (MDS-EB-1) or type 2 (MDS-EB-2) • MDS, unclassifiable (MDS-U) |
| International Consensus Classification (2022)17 | MDS subtypes • MDS with mutated SF3B1 (MDS-SF3B1) • MDS with del(5q) (MDS del(5q)) • MDS, NOS—without dysplasia • MDS, NOS—with single-lineage dysplasia • MDS, NOS—with multilineage dysplasia • MDS with excess blasts (MDS-EB) • MDS with mutated TP53 • MDS/AML |
| Related clonal disorders • Clonal hematopoiesis of indeterminate potential (CHIP) • Clonal cytopenia of undetermined significance (CCUS) • VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome: autoinflammatory syndrome associated with anemia and clonal hematopoiesis due to somatic mutation in the UBA1 gene | |
| 2022 WHO classification (fifth edition)20 | MDS with defining genetic abnormalities • MDS with low blasts and isolated 5q deletion (MDS-5q) • MDS with low blasts and SF3B1 mutation (MDS-SF3B1) • MDS with biallelic TP53 inactivation (MDS-bi TP53) MDS, morphologically defined • MDS with low blasts (MDS-LB) • MDS, hypoplastic (MDS-h) • MDS with increased blasts (MDS-IB, including MDS with fibrosis) |
NOS, not otherwise specified.
The WHO subtypes are associated with different risks of death from complications of cytopenia or evolution to AML,3 while cytogenetic abnormalities are an additional independent prognostic factor in MDS.4 The Revised International Prognostic Scoring System (IPPS-R) considers hemoglobin level, platelet count, absolute neutrophil count, cytogenetic abnormalities, and bone marrow blasts.5 The IPSS-R defines 5 risk categories (very low, low, intermediate, high, and very high risk) and is currently universally used for risk stratification in MDS.1,5 In clinical practice, a cutoff of 3.5 for the IPSS-R score defines 2 distinct risk groups: lower-risk MDS (score ≤3.5, median survival 5.9 years) and higher-risk MDS (score >3.5, median survival 1.5 years).6 This risk stratification so far has been the basis of a patient-centered care approach to the treatment of MDS.1
CLINICAL CASE 1
A 60-year-old man is evaluated for mild exertional dyspnea. A complete blood count (CBC) shows the following: hemoglobin (Hb), 10 g/dL; platelet (PLT) count, 240 × 109/L; white blood cell (WBC) count, 3.8 × 109/L; and absolute neutrophil count (ANC), 2.1 × 109/L. The patient is seen by a hematologist who uses the 2016 revised WHO classification of MDS and the IPSS-R. A bone marrow examination reveals erythroid dysplasia, 65% ring sideroblasts, and 1% blasts, while cytogenetics is normal. A diagnosis of MDS with ring sideroblasts is made; the IPSS-R risk score is 1 and the risk category is very low with an expected median survival of approximately 9 years. The condition is clinically defined as lower-risk MDS, and a medical treatment (an erythropoiesis-stimulating agent first, luspatercept as a second-line treatment in case of transfusion requirement) is planned.1
CLINICAL CASE 2
A 66-year-old woman is evaluated for fatigue. A CBC shows the following: Hb, 10 g/dL; PLT count, 95 × 109/L; WBC count, 4.0 × 109/L; and ANC, 1.5 × 109/L. The patient is seen by a hematologist who uses the 2016 revised WHO classification of MDS and the IPSS-R. A bone marrow examination reveals multilineage dysplasia with 2% blasts; cytogenetics shows isolated del(7q) (intermediate cytogenetic risk group). A diagnosis of MDS with multilineage dysplasia is made; the IPSS-R risk score is 2.5 and the risk category is low with an expected median survival of 5.3 years.1 The condition is clinically defined as lower-risk MDS, and a medical treatment (an erythropoiesis-stimulating agent) is planned.1
MDS in the genomics era
The beginning of the genomics era in medicine has been marked by the introduction of massively parallel DNA sequencing methods, commonly known as next-generation sequencing.7 The use of these methods in well-characterized patient populations has clarified the genetic basis of MDS and has enabled the development of novel approaches to diagnosis and prognostication for patients with these neoplasms.
Genetic basis of MDS
MDS arises from the growth and spread of a somatically mutated clone of hematopoietic cells.1 Several mutation-driver genes can cause clonal proliferation; biologic pathways include RNA splicing, DNA methylation, histone modification, transcription regulation, DNA repair control, signaling, and cohesin complex.8,9 Most patients have combinations of pathway mutations; at clinical onset, the median number of driver mutations is 2 to 3 per patient.10 The combination of bone marrow hyperplasia and peripheral blood cytopenia represents the paradox of myelodysplastic hematopoiesis.1 Mutation-driver genes provide an advantage at the hematopoietic stem cell and progenitor cell level, determining clonal proliferation, but this is associated with a disadvantage at the hematopoietic precursor level, leading to ineffective hematopoiesis and peripheral blood cytopenia.
Leukemic transformation
Evolution to AML occurs through a process of clonal selection and may follow different patterns.11 For instance, SF3B1- mutant MDS is a relatively indolent disorder with a long-lasting chronic phase characterized by isolated anemia12 ; in a minority of patients, acquisition or expansion of a somatic mutation in RUNX1 can eventually determine leukemic transformation.12 By contrast, most patients with multiple hits in TP53—TP53 multihit state consistent with biallelic targeting and inactivation— rapidly progress to AML, indicating a continuum between myelodysplastic and leukemic phase in the condition.13
Germline predisposition
In the past few years, several studies have shown that more than 10% of patients with MDS have a genetic predisposition to myeloid neoplasms.14 In these individuals, the somatic driver mutation that causes myelodysplastic proliferation occurs on the background of a germline variant. A list of conditions with germline predisposition to myeloid neoplasms is reported in Table 2.
Conditions with germline predisposition to myeloid neoplasm that may progress to MDSa
| Conditions without clinical manifestations before the development of a myeloid neoplasm |
|---|
| • DDX41-associated susceptibility to myeloid malignancies |
| • Li-Fraumeni syndrome, associated with germline TP53 mutations |
| Constitutional platelet disorders, characterized by thrombocytopenia and/or platelet dysfunction |
| • RUNX1-related familial platelet disorder |
| • ANKRD26-related thrombocytopenia |
| • ETV6-related thrombocytopenia |
| Inherited bone marrow failure syndromes |
| • Fanconi anemia |
| • Diamond-Blackfan anemia |
| • Shwachman-Diamond syndrome |
| • Dyskeratosis congenita (X-linked telomere disease) |
| • Other telomerase biology disorders |
| Miscellaneous syndromes featuring abnormalities of hematopoiesis and other organs |
| • GATA2-spectrum disorders (GATA2 deficiency syndrome) |
| • SAMD9-related MIRAGE syndrome |
| • SAMD9L-related ataxia-pancytopenia |
| • ERCC6L2-associated bone marrow failure syndrome |
| Conditions without clinical manifestations before the development of a myeloid neoplasm |
|---|
| • DDX41-associated susceptibility to myeloid malignancies |
| • Li-Fraumeni syndrome, associated with germline TP53 mutations |
| Constitutional platelet disorders, characterized by thrombocytopenia and/or platelet dysfunction |
| • RUNX1-related familial platelet disorder |
| • ANKRD26-related thrombocytopenia |
| • ETV6-related thrombocytopenia |
| Inherited bone marrow failure syndromes |
| • Fanconi anemia |
| • Diamond-Blackfan anemia |
| • Shwachman-Diamond syndrome |
| • Dyskeratosis congenita (X-linked telomere disease) |
| • Other telomerase biology disorders |
| Miscellaneous syndromes featuring abnormalities of hematopoiesis and other organs |
| • GATA2-spectrum disorders (GATA2 deficiency syndrome) |
| • SAMD9-related MIRAGE syndrome |
| • SAMD9L-related ataxia-pancytopenia |
| • ERCC6L2-associated bone marrow failure syndrome |
Information is from Cazzola.1
The International Consensus Classification of MDS: increasing impact of genomics
The revised fourth edition of the WHO classification of hematologic neoplasms was developed in 2016 based on input from clinical advisory committees composed of pathologists, clinicians, and genomic scientists.2 In 2021, a group of these pathologists and clinicians decided to independently proceed with a clinical advisory committee effort by organizing the International Consensus Conference on the classification of myeloid and lymphoid neoplasms.15 This led to the development of the International Consensus Classification of myeloid and lymphoid neoplasms, which is described in 2 Special Reports in Blood.16,17
The International Consensus Classification of MDS is reported in Table 1: of note, 3 MDS subtypes are defined by genomic features. One of them is MDS with isolated del(5q), which had previously been established as a distinct nosologic entity.15 The 5q32-q33 deletion is responsible for the haploinsufficiency production of multiple gene transcripts, which explains the clonal proliferation, the macrocytic anemia, and the efficacy of lenalidomide treatment.1 In addition, the International Consensus Classification of MDS includes 2 new nosologic entities that are defined by somatic gene mutations. The first one is SF3B1- mutant MDS, characterized by persistent cytopenia, no excess blasts, and SF3B1 mutation (with a variant allele frequency [VAF] ≥10%) without any concomitant mutation in RUNX1 or a TP53 multihit state. SF3B1-mutant MDS has a relatively indolent clinical course; in fact, patients with isolated SF3B1 mutation have a median overall survival of approximately 9 years, with a very low risk of leukemic evolution.12 The second MDS subtype defined by molecular features is MDS with mutated TP53, characterized by cytopenia, less than 10% bone marrow blasts, and TP53 multihit state or TP53 mutation (VAF ≥10%) plus complex karyotype. TP53 multihit patients have a median overall survival of approximately 9 months with a very high risk of leukemic evolution.13
In addition to the abovementioned, molecularly defined MDS subtypes, the International Consensus Classification of MDS includes other conditions whose diagnosis requires molecular profiling. One of these is clonal cytopenia of undetermined significance, characterized by unexplained cytopenia in 1 or more peripheral blood cell lineages; a somatic mutation, at a VAF of at least 2%, in 1 or more genes that are recurrently mutated in myeloid neoplasms; and insufficient criteria for a diagnosis of MDS.1,17 The other condition is the VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome, a unique autoinflammatory syndrome associated with anemia and clonal hematopoiesis due to somatic mutation in the UBA1 gene.18 The International Consensus Classification recommends keeping VEXAS separate from MDS, unless morphologic criteria of MDS are met, typically in the setting of additional somatic genetic lesions.17
The new WHO classification of MDS
On 22 June 2022, 2 review articles covering the fifth edition of the WHO classification of myeloid and lymphoid neoplasms were published online.19,20 At the time of writing, the related Blue Book monograph has not been published.
The updated WHO classification of MDS—renamed as myelodysplastic neoplasms—is shown in Table 1. The subtypes defined by genomic features are essentially the same as in the International Consensus Classification. Hypoplastic MDS21 has been recognized as a distinct disease type, while MDS with fibrosis22 has been included within MDS with increased blasts.
Risk stratifying with the International Prognostic Scoring System–Molecular
The IPSS-R is universally used not only for risk stratification and therapeutic decision-making in the clinic but also in designing clinical trials; in turn, it is included in the approved indications (drug labels) of drug regulatory agencies. A clear limitation of the IPSS-R in the genomics era is that it considers cytogenetic abnormalities but not somatic gene mutations, which have been shown to have independent prognostic significance.1 This limitation and the need for molecular precision were discussed recently in this journal.23-25 Recent proof-of-concept studies have underlined the independent prognostic significance of somatic gene mutations but have unfortunately failed to provide any clinically useful prognostic tool.26-28
Intending to reach molecular precision, the International Working Group for Prognosis in MDS (IWG-PM) has developed the International Prognostic Scoring System–Molecular (IPSS-M) by studying a well-characterized cohort of 2957 patients with MDS who were profiled for mutations in 152 genes.29 Patients were recruited from 24 centers in different regions of the world. All were treatment naive at diagnosis and not selected on the basis of the therapies they received during their clinical course. As such, 30% of patients were actively treated per standard of care. Overall survival was available for 95% of patients and leukemia-free survival for 88%; the median follow-up was 3.8 years. Data from 754 Japanese patients with MDS were used for validation.
SF3B1 was the only gene consistently associated with favorable outcomes across all the above end points, while TP53 multihit state was one of the top predictors of adverse outcomes. The number of molecular abnormalities per patient correlated with the severity of clinical outcomes, and comutation patterns had a significant impact. For instance, patients with the SF3B1 mutation as a sole genetic lesion had a median overall survival of approximately 9 years with a negligible risk of leukemic transformation, but those with concomitant SF3B1 and RUNX1 mutation had a significantly lower leukemia-free and overall survival.29
The IPSS-M risk score was built as a continuous index, defined as a weighted sum of the following prognostic variables: Hb, PLT count, bone marrow blast percentage, IPSS-R cytogenetic category, 17 binary features derived from the presence of mutations in 16 prognostic genes, and a feature representing the number of mutated genes (Nres) from a residual group of 15 genes. These variables were evaluated for associations with leukemia-free survival (primary end point), leukemic transformation, and overall survival.
The IPSS-M risk score was scaled so that a score value of 0 represented the average patient, that is, a hypothetical patient with mean values for all variables. Negative values indicate lower risk, while positive values denote higher risk. A change in score of +1 is associated with a doubling of risk, while a change of −1 involves a halving of risk. While the score is patient specific, to facilitate the use of the IPSS-M in the clinic, a 6-risk category schema was defined based on score cutoffs: the different risk groups and their clinical outcomes are summarized in Table 3.29 Based on the C-index, the IPSS-M resulted in improved discrimination compared to the IPSS-R score.
IPSS-M risk categories and clinical outcomesa
| Risk category | Median leukemia-free survival, y | Median overall survival, y | AML transformation by 1 year, % |
|---|---|---|---|
| Very low (14% of all patients) | 9.7 | 10.6 | 0 |
| Low (32%) | 5.9 | 6.0 | 1.7 |
| Moderately low (11%) | 4.5 | 4.6 | 4.9 |
| Moderately high (11%) | 2.3 | 2.8 | 9.5 |
| High (14%) | 1.5 | 1.7 | 14.3 |
| Very high (18%) | 0.7 | 1.0 | 28.2 |
| Risk category | Median leukemia-free survival, y | Median overall survival, y | AML transformation by 1 year, % |
|---|---|---|---|
| Very low (14% of all patients) | 9.7 | 10.6 | 0 |
| Low (32%) | 5.9 | 6.0 | 1.7 |
| Moderately low (11%) | 4.5 | 4.6 | 4.9 |
| Moderately high (11%) | 2.3 | 2.8 | 9.5 |
| High (14%) | 1.5 | 1.7 | 14.3 |
| Very high (18%) | 0.7 | 1.0 | 28.2 |
Information is from Bernard et al.29
The IWG-PM has built the IPSS-M web-based calculator (MDS Risk Support: https://mds-risk-model.com), which returns not only the individual score but also the expected leukemia-free survival, overall survival, and risk of AML transformation. The calculator accounts for missing values; in these cases, the IPSS-M is calculated under the best, average, and worst scenarios.
Future directions in genomic profiling for clinical decision-making in MDS
The available evidence indicates that gene sequencing substantially improves the diagnostic process and risk stratification in MDS.1 Despite this, how to implement genomic profiling for clinical decision-making is still a debated question.30 Chromosome banding analysis complemented by gene panel sequencing is already a routine procedure at most academic institutions in the United States and Western Europe. However, gene copy number and loss of heterozygosity assessment, which are essential for recognition of the TP53 multihit state,13,29 are not routinely included in gene panel sequencing.
Duncavage et al31 have recently shown that whole-genome sequencing represents a potential replacement for both conventional cytogenetic and sequencing approaches in patients with MDS, providing rapid and accurate comprehensive genomic profiling. These findings have been independently confirmed by Haferlach et al,32 who concluded that whole-genome sequencing will likely outperform gene panel sequencing in a few years, especially in entities with a high level of genetic complexity. Furthermore, the feasibility of whole-genome and transcriptome profiling has been recently demonstrated in pediatric cancers.33 Provided that ad hoc infrastructures are built in our health systems, whole-genome sequencing will likely become a routine method for genomic profiling in myeloid malignancies in the next years.7
Molecular profiling should not focus exclusively on somatic mutations. Germline predisposition to myeloid neoplasms now includes a plethora of heterogeneous conditions that may have no or mild clinical manifestations before the progression to myeloid malignancy (Table 2).34 For instance DDX41-associated susceptibility to myeloid malignancies does not involve any clinical phenotype in the first 5 to 6 decades of life. These individuals carry a heterozygous germline mutation in DDX41; acquisition of a somatic mutation in the normal DDX41 allele (or in another myeloid gene) may lead to MDS in the sixth or seventh decade of life.35,36 Although individual predisposition disorders are relatively rare, collectively they likely account for more than 10% of all cases of MDS.1 In a recent study, pathogenic germline variants were identified in 28 of 404 (7%) patients with MDS of all ages treated with allogeneic stem cell transplantation from a family donor.37 Because variants were called as those detectable in both the transplanted patient and related donor, the 7% overall frequency of deleterious germline variants is clearly an underestimate.
At present, germline testing is considered in patients with MDS younger than 50 years, in those with syndromic or suggestive clinical features, or in those with a suggestive family history. In perspective, however, germline testing should be considered in patients with MDS.
A potential future approach to diagnosis and risk stratification of MDS is illustrated in Figure 1.
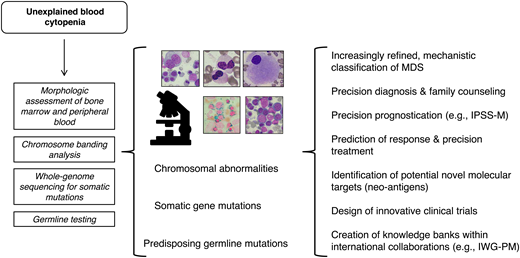
A future approach to the diagnosis and risk stratification of MDS that will enable precision treatments. To make this approach feasible, ad hoc infrastructures should be built into our health systems.7
A future approach to the diagnosis and risk stratification of MDS that will enable precision treatments. To make this approach feasible, ad hoc infrastructures should be built into our health systems.7
CLINICAL CASE 1 (Continued)
A 60-year-old man is evaluated for mild exertional dyspnea. A CBC shows the following: Hb, 10 g/dL; PLT count, 240 × 109/L; WBC count, 3.8 × 109/L; and ANC, 2.1 × 109/L. The patient is seen by a hematologist who uses the International Consensus Classification of MDS and the IPSS-M. A bone marrow examination reveals erythroid dysplasia, 65% ring sideroblasts, and 1% blasts; cytogenetics is normal. Panel gene sequencing detects an SF3B1 mutation (VAF 45%) combined with a subclonal RUNX1 mutation (VAF 3%). A diagnosis of MDS with single-lineage (erythroid) dysplasia, somatic driver mutations in SF3B1, and subclonal mutation in RUNX1 is made. The IPSS-M risk score is −0.96, and the risk category is low with an expected median survival of 6 years. While a medical treatment is planned, the patient's eligibility for allogeneic stem cell transplantation is also evaluated, primarily because the RUNX1-mutant subclone might expand over time and drive leukemic transformation.1,12 The patient is followed regularly (3 times per year) and monitored not only for CBC but also for the RUNX1-mutation VAF; a significant increase may represent an indication for proceeding to allogeneic transplantation if the patient is eligible.
CLINICAL CASE 2 (Continued)
A 66-year-old woman is evaluated for fatigue. A CBC shows the following: Hb, 10 g/dL; PLT count, 95 × 109/L; WBC count, 4.0 × 109/L; and ANC, 1.5 × 109/L. The patient is seen by a hematologist who uses the International Consensus Classification of MDS and the IPSS-M. A bone marrow examination reveals multilineage dysplasia with 2% blasts; cytogenetics shows isolated del(7q) (intermediate cytogenetic risk group). Panel gene sequencing detects a TP53 multihit state. A diagnosis of MDS with mutated TP53 is made; the IPSS-M risk score is 1.12 and the risk category is high with an expected median survival of 1.7 years and a high risk of leukemic transformation. The clinician evaluates the patient's eligibility for allogeneic stem cell transplantation and considers the patient's enrollment into an innovative clinical trial, as MDS with mutated TP53 has a poor outcome with either hypomethylating agents or allogeneic transplantation.13
Conclusions
Our understanding of the molecular pathophysiology of MDS has improved remarkably in recent years, and this has provided novel diagnostic/prognostic approaches.1 The recently developed International Consensus Classification and the 2022 WHO classification of MDS include new entities that are molecularly defined—namely, SF3B1-mutant and TP53-mutant MDS, in addition to the already established MDS with isolated del(5q). In addition, the IPSS-M Risk Calculator for MDS is currently available online.29
Nonetheless, a lot of work remains to be done regarding the classification of MDS, which is still largely based on morphologic criteria. For instance, the existing classifications do not specifically consider the spliceosome genes SRSF2 and U2AF1, which are mutated in MDS subtypes characterized by a high level of genetic complexity and poor clinical outcome.1 Also, the current classifications do not consider the epigenetic regulators DNMT3A, TET2, and ASXL1, collectively called DTA; as a sole genetic lesion, DTA genes are associated with MDS subtypes with relatively good clinical outcomes.
To develop an increasingly mechanistic classification of MDS, a collective effort is needed, as discussed in a recent perspective article.38 The knowledge bank created by the IWG-PM, which has enabled the development of the IPSS-M, represents a straight example of independent, productive collaboration.29 Ongoing investigations based on this knowledge bank will likely lead to a genetically inspired classification of MDS.
Acknowledgments
I am profoundly grateful to all patients I have met in the past decades, to the Associazione Italiana per la Ricerca sul Cancro (AIRC, Milan, Italy) that funded our investigations in MDS, and to the many friends I have collaborated with under the aegis of the MDS Foundation.
Conflict-of-interest disclosure
Mario Cazzola declares no potential conflict of interest.
Off-label drug use
Mario Cazzola: nothing to disclose.
