

Abstract
Gastrointestinal (GI) bleeding is an important cause of morbidity and mortality in von Willebrand disease (VWD). It has been noted that GI bleeding caused by angiodysplasia is overrepresented in VWD patients compared to other causes. The bleeding from angiodysplasia is notoriously difficult to treat; recurrences and rebleeds are common. A growing body of basic science evidence demonstrates that von Willebrand factor negatively regulates angiogenesis through multiple pathways. VWD is clinically highly associated with angiodysplasia. The predisposition to angiodysplasia likely accounts for many of the clinical difficulties related to managing GI bleeding in VWD patients. Diagnosis and treatment are challenging with the current tools available, and much further research is needed to further optimize care for these patients with regard to acute treatment, prophylaxis, and adjunctive therapies. In this review we provide an overview of the available literature on GI bleeding in VWD and explore the molecular underpinnings of angiodysplasia-related GI bleeding. Considerations for diagnostic effectiveness are discussed, as well as the natural history and recurrence of these lesions and which therapeutic options are available for acute and prophylactic management.
Learning Objectives
Teach the investigation and management of GI bleeding in VWD patients
Describe the role that angiodysplasia plays in VWD
CLINICAL CASE
A 59-year-old male with type 2B VWD presents to the hospital with a 1-day history of melena stools. During previous bleeding events, he usually received treatment on demand with von Willebrand factor (VWF) concentrate and tranexamic acid (TXA). Upon examination, he is vitally stable. The lab work is notable for a hemoglobin of 85 g/L (compared to 140 g/L at his last clinic visit 2 months ago). The patient is admitted to the hospital and treated with a VWF/factor VIII (FVIII) concentrate of 80 IU/kg intravenously every 8 hours. Central venous access is obtained to assist with resuscitation. Upper and lower endoscopies are performed the next day and do not show the source of the bleeding. The patient then undergoes a VCE study that demonstrates a bleeding angiodysplasia lesion in the ileum. The lesion is treated with argon plasma coagulation. The patient is titrated down to daily VWF/FVIII prophylaxis over the next week. Ten days after the initial admission, the patient, with stable hemoglobin and no clinical evidence of bleeding, is discharged home on daily prophylaxis.
Background
VWF acts as a bridging molecule for platelet adhesion and as a carrier for FVIII during normal hemostasis. Under high shear conditions, VWF also promotes platelet aggregation.1 Defects or deficiencies in VWF occur in VWD, which is the most common inherited bleeding disorder. VWD has an overall prevalence in the general population as high as 1%, with symptomatic disease occurring in 0.1%.2 Further classification of VWD is based on the type of deficiency. VWD type 1 occurs in 75% of cases and is a partial quantitative deficiency resulting in the mildest VWD phenotype.3 VWD type 2 is characterized by qualitative deficiencies of VWF and has multiple subclassifications. The type 2A subtype has a selective deficiency of high-molecular-weight multimers (HMWM) of VWF due to either impaired multimerization or enhanced proteolysis. In contrast, in type 2B VWD there is also reduced HMWM as well as variable thrombocytopenia; however, the mechanism is due to the enhanced affinity of VWF for the platelet glycoprotein Ib (GPIb) receptor as a result of gain-of-function mutations, reducing the quantities of both binding partners. Subtypes 2M and 2N are less common and are the result of variants that make VWF less effective at binding to GPIb and FVIII, respectively. Type 3 VWD reflects a near complete deficiency of VWF, is the rarest of the classifications, and has the most serious clinical manifestations.4,5
Unsurprisingly, the primary clinical manifestation and presentation of VWF deficiency leading to diagnosis is bleeding. Bleeding primarily presents as gastrointestinal (GI) bleeding, epistaxis, or menorrhagia. Heavy menstrual bleeding is the most common initial presentation. While GI bleeding occurs less frequently than epistaxis or menorrhagia, it carries a higher morbidity and mortality.6 Notably, GI bleeding is the most common reason for VWD patients to require hospitalization during a bleeding episode.7 In this review we discuss the pathophysiology of GI bleeding in VWD, particularly with respect to angiodysplasia, outline optimal diagnostic strategies, and explore treatment options.
VWD and angiodysplasia
Clinically, it has been demonstrated that qualitative or quantitative defects of VWF are associated with vascular malformations, especially in the GI tract.8 This occurs in both congenital VWD and acquired von Willebrand syndrome (AVWS). In one of the first large-scale descriptions of this phenomenon via a survey conducted in the 1990s, the incidence of vascular malformations only occurred in VWD patients with absent HMWMs of VWF. A study comparing bleeding tendencies between VWD type 2A and 2M, which have similar VWF and FVIII levels but differ in HMWM loss, found that bleeding rates were higher in incidence and severity among type 2A patients.9 While this lack of HMWM was initially suggested as the causative mechanism, subsequent studies have found angiodysplasia overrepresented in all other VWD subtypes, suggesting other mechanisms may also contribute.10 Angiodysplasia prevalence and bleeding rates differ between VWD subtypes. A scoping review of the available literature found that type 2A represented 39% of reported cases and type 3, 14.4%, while type 1 made up only 11.6%.11 Population-level data on prevalence are lacking due to reports overwhelmingly being case reports.
Angiodysplasia in VWD also occurs outside the GI tract, although most (>85%) is gastrointestinal.11 Interestingly, in a scoping review of vascular abnormalities in VWD patients, 10% of the 146 patients included had a concurrent diagnosis of hereditary hemorrhagic telangiectasia. A prior study evaluating the microcirculation in 100 VWD patients' fingers using intravital capillary microscopy to visualize the nail bed demonstrated increased dilatation, microscopic bleeding, and dysplastic changes. These abnormalities were highly predictive of the diagnosis of VWD as opposed to other bleeding disorders, such as hemophilia.12 While angiodysplasia can be present in multiple locations, the GI tract deserves special note due to the potential to cause serious illness from bleeding complications.
Molecular mechanisms of angiodysplasia in VWD
The precise underlying mechanisms of angiodysplasia in VWD have been explored in cellular and animal models. Increased vascular endothelial growth factor (VEGF)–dependent angiogenesis was recently documented in endothelial cells (ECs) and mice deficient in VWF.13,14 These results provide direct evidence of the regulatory role of VWF in angiogenesis and a connection to angiodysplasia found in patients with absent or abnormal VWF. Subsequent studies in this area have sought to further investigate the molecular pathways by which VWF regulates angiogenesis, taking into consideration the numerous binding partners of VWF and the many possible mechanisms through which VWF may regulate vessel formation.15 The current evidence suggests that VWF modulates angiogenesis via both the extracellular and the intracellular pathways that promote VEGF signaling (Figure 1).13,15
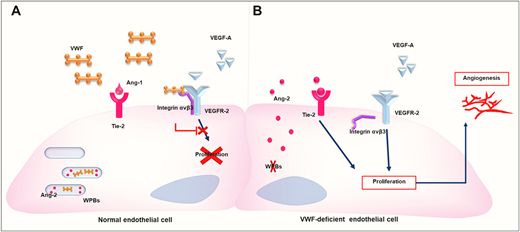
Molecular mechanisms of angiodysplasia driven by abnormalities in VWD based on comparisons between normal cells (A) and VWF-deficient ECs (B). Adapted with permission from Selvam and James21 and Allerkamp et al.22
In the extracellular pathway, VWF modulates angiogenesis by interacting with integrin αvβ3. It is suggested that the binding of VWF to ECs via integrin αvβ3 results in the inhibition of VEGFR-2 signaling and the subsequent inhibition of angiogenesis.16 It has also been observed that the absence of integrin αvβ3 in mice is associated with increased VEGFR-2–dependent angiogenesis.17 Similar enhanced VEGFR-2–dependent angiogenesis has been observed in VWF-deficient ECs.13 Interestingly, the presence of extracellular VWF only partially inhibited the angiogenic potential of VWF-deficient human umbilical vein ECs, suggesting that an intracellular VWF pathway may be involved in regulating vessel formation.13
In the intracellular pathway, the absence of Weibel-Palade bodies, which are storage organelles for VWF and other bioactive molecules, either due to defective or deficient VWF, results in the free release of angiopoietin-2 (Ang-2) from ECs. This is proposed to promote EC angiogenic activity via Tie2 receptors.13,18 VWF is known to promote the formation of Weibel- Palade bodies, which store several molecules, including VWF and Ang-2.19 In fact, VWF has been shown to regulate the synthesis, storage, and release of Ang-2, with increased mRNA levels of Ang-2 noted in VWF-deficient mice and individuals with type 3 VWD.14
Previous studies showing that Ang-2 plays a role in the internalization and degradation of integrin αvβ3 confirm an interconnection between the two pathways.20 Additionally, both the extracellular and intracellular pathways make use of VEGFR-2 signaling, and thus a deficiency of VWF results in increased VEGFR-2–mediated angiogenesis.13 VWF, therefore, regulates angiogenesis through VEGFR-2 signaling inhibition. Interestingly, multiple studies have implicated abnormally increased VEGFR-2 signaling in the development of fragile and leaky vessels, as observed in VWD patients.15
VWD, angiodysplasia, and other causes of GI bleeding
In a survey of patients admitted to US hospitals for GI bleeding, angiodysplasia occurred in 36.5% of cases with a VWD diagnosis. This was significantly higher than the non-VWD patients with GI bleeding, 9.5% of whom were hospitalized as a result of angiodysplasia.23 The true prevalence of angiodysplasia in VWD patients is likely underestimated due to diagnostic difficulties. A study of 48 VWD patients with a history of unexplained GI bleeding who had already completed conventional endoscopy and systematic VCE evaluation revealed that 38% had previously undiagnosed GI angiodysplasia.24 Interestingly, GI bleeding from other causes, such as colon cancer or peptic ulcer disease, appears to occur at similar rates in VWD patients as the general population, although due to underlying hemostatic defects, the bleeds are more severe.25
Investigations into VWD rates in patients with angiodysplasia and the prevalence of angiodysplasia in VWD are limited but suggest that up to 10% of patients with angiodysplasia may have a VWD diagnosis.11 Overall, VWD remains overrepresented among patients with angiodysplasia, and angiodysplasia is a significant cause of bleeding in VWD. These findings are especially striking when considering that the prevalence of angiodysplasia in non-VWD patients is reported as less than 1%.26 Evaluating for VWD or acquired von Willebrand syndrome in patients with angiodysplasia should be considered. A previous systematic evaluation of 9 patients with bleeding angiodysplasias found abnormal VWF, particularly HMWM deficiency, in 8 of those patients. However, all 8 patients had a detectable reason for AVWS, and congenital VWD was not diagnosed.27 Given the advances in the diagnosis of both GI bleeding and VWD, further prospective work is needed in evaluating VWF levels and function in angiodysplasia patients.
Diagnosis of the cause of GI bleeding in VWD
Since the source of GI bleeding in VWD is often from angiodysplasia, diagnosis can be challenging. VWD patients are most commonly diagnosed with angiodysplasia by conventional endoscopy or VCE.11 Conventional upper and lower endoscopies are the initial tests of choice, primarily due to easier access rather than superior testing characteristics. Conventional endoscopy is also important to rule out other causes of GI bleeding, such as ulcers or malignancy. Given the potential for a life-threatening bleed, investigations and management should be concurrent.
Both VCE and enteroscopy (using an endoscope to visualize the small bowel) allow visualization of the small bowel, a common location for angiodysplasia in VWD patients. Visualization in enteroscopy is limited compared to VCE but has the benefit of the option of concurrent therapeutic intervention at the time of visualization.28 Even with VCE, more than 20% of GI bleeding in VWD patients does not have a clear source.29 Importantly, even if angiodysplasia is not the underlying diagnosis, the ability to diagnose alternate causes of GI bleeding in VWD patients improves when VCE is performed in addition to conventional endoscopy.24,29
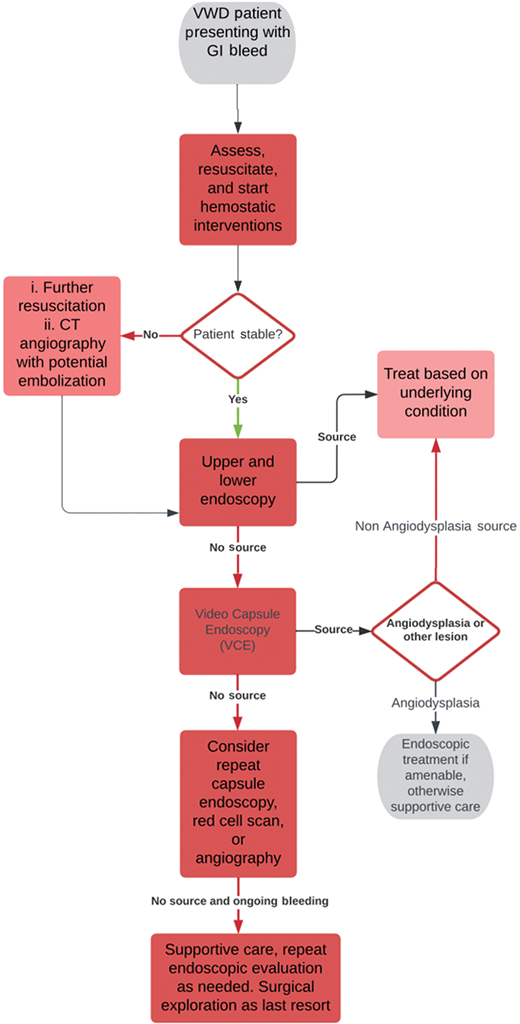
VCE has limitations. Lesions that are not actively bleeding may be more difficult to visualize and may remain unidentified. There is also an approximately 2% risk of a retained capsule—patients with VWD likely have multiple studies and may have higher complication rates.30 Other diagnostic studies, such as angiography or a red cell scan, can be beneficial in certain circumstances but require a minimum volume of ongoing blood loss to detect a significant finding, and consequently, in angiodysplasia related bleeding is often unrevealing.31 Overall, diagnosis is challenging and often made by exclusion. A suggested workup that incorporates information from existing guidelines on GI bleeding is shown in Figure 2.31,32
Treatment of GI bleeding in VWD—acute management
The treatment of acute GI bleeding for VWD patients is like their other bleeding events and involves correcting the primary hemostatic defect from a lack of VWF and increasing FVIII. In addition to standard therapies such as desmopressin, VWF concentrate, and factor replacement,3 endoscopic and procedural interventions can provide definitive management (Table 1). Generally, GI bleeding is more difficult to control than other types of bleeds, and double the VWF concentrate is needed to achieve hemostasis in retrospective evaluations.7 There are no specific recommendations as to what product should be utilized in these patients with respect to choosing products containing VWF alone or products combining VWF and FVIII (each with a variable ratio of the two components). Evidence with newer recombinant VWF concentrates, such as Vonicog Alfa, in GI bleeding is limited but potentially promising.33 Vonicog Alfa has an abundance of ultra-high weight multimers and is worthy of further study in VWD-related GI bleeding, particularly if caused by angiodysplasia.
Acute hemostatic and endoscopic interventions for GI bleeding in VWD
| Intervention | Dosage | Mechanism | Cautions | Notes |
|---|---|---|---|---|
| Desmopressin3 | 0.3 µg/kg with repeat doses given as needed | Increases plasma levels of VWF and FVIII | Contraindicated in type 2B due to increased platelet binding casing thrombocytopenia; ineffective in type 3 | Patients should have established response with desmopressin challenge |
| VWF/FVIII concentrate3 | Dependant on indication, more severe bleeds have higher suggested dose. | Exogenously increases levels of VWF and FVIII | Potential for inhibitor development, infusion reactions | Suggested targets of factor VIII >50 IU/dL and VWF:RCo >50 IU/dL |
| TXA3 | Dose varies: suggested 1-2 g IV loading dose and up to 1 g IV every 8 hours afterward until hemostasis achieved34,36 | Antifibrinolytic agent | Avoid if hematuria present | |
| Argon plasma coagulation28 | N/A | Electric current conducted through argon gas | Depth of coagulation only a few millimeters | |
| Embolization31 | N/A | Occlusion of culprit vessel | Greater risk of complications, including ischemic bowel | Only effective if source of bleed comes from a specific blood vessel |
| Surgical resection24 | N/A | Removal of affected GI tract | Unlikely to resolve all bleeding if further angiodysplasias develop | Last resort, high complication rate |
| Electrocautery8–28 | N/A | Tissue and vessel destruction with direct application of electric current | Risk of perforation higher than argon plasma coagulation |
| Intervention | Dosage | Mechanism | Cautions | Notes |
|---|---|---|---|---|
| Desmopressin3 | 0.3 µg/kg with repeat doses given as needed | Increases plasma levels of VWF and FVIII | Contraindicated in type 2B due to increased platelet binding casing thrombocytopenia; ineffective in type 3 | Patients should have established response with desmopressin challenge |
| VWF/FVIII concentrate3 | Dependant on indication, more severe bleeds have higher suggested dose. | Exogenously increases levels of VWF and FVIII | Potential for inhibitor development, infusion reactions | Suggested targets of factor VIII >50 IU/dL and VWF:RCo >50 IU/dL |
| TXA3 | Dose varies: suggested 1-2 g IV loading dose and up to 1 g IV every 8 hours afterward until hemostasis achieved34,36 | Antifibrinolytic agent | Avoid if hematuria present | |
| Argon plasma coagulation28 | N/A | Electric current conducted through argon gas | Depth of coagulation only a few millimeters | |
| Embolization31 | N/A | Occlusion of culprit vessel | Greater risk of complications, including ischemic bowel | Only effective if source of bleed comes from a specific blood vessel |
| Surgical resection24 | N/A | Removal of affected GI tract | Unlikely to resolve all bleeding if further angiodysplasias develop | Last resort, high complication rate |
| Electrocautery8–28 | N/A | Tissue and vessel destruction with direct application of electric current | Risk of perforation higher than argon plasma coagulation |
IV, intravenously; N/A, not applicable; VWF:RCo, VWF ristocetin cofactor activity.
Adjunctive therapies used during an acute bleed, such as TXA, should be administered in VWD-related bleeds. One evaluation showed a high rate of response (55%) with TXA, but patients often receive multiple therapies.24 Of note, a recent large randomized controlled trial of TXA in non-VWD GI bleeds, the HALT-IT trial, demonstrated no benefit and some evidence of harm. However, TXA is still suggested in VWD.34 As with other GI bleeds, anticoagulant and antiplatelet agents should be withheld, with consideration for reversal if these products are available and indicated.
Endoscopic treatment should not be neglected, and the timely involvement of the gastroenterology department for diagnosis and management is imperative. Treatment endoscopically is based on the underlying cause of the bleed. Angiodysplasia deserves special consideration due to its prevalence among VWD patients. Unfortunately, because angiodysplasia typically occurs in the small bowel, it is often not therapeutically accessible via traditional endoscopic methods.28 Endoscopic options include argon plasma coagulation and bipolar electrocoagulation. These are successful for angiodysplasia when there is a single lesion or only a few small lesions; however, complication rates increase with the number of interventions. Laser treatment, for example, has a higher perforation rate than argon plasma coagulation but provides hemostasis to a deeper depth.28 In extreme circumstances, surgical management that includes resection of an entire section of bowel could be completed, but success is variable if there is no clear source (such as a malignancy).11,35 If the source is angiodysplasia, the operation may be futile since more angiodysplasia will develop over time, given that the underlying disease drives its ongoing development.
Prophylaxis for GI bleeding in VWD
Prophylactic treatment with VWF concentrate is the most effective method of preventing recurrent bleeding. In patients with VWD with a history of severe bleeds, prophylaxis is suggested by the most recent guidelines.3 The VWD Prophylaxis Network study is the largest-known registry study designed to evaluate prophylaxis outcomes.37 Among patients with severe bleeding from VWD, about a quarter in the study were started on prophylaxis for GI bleeding. Prophylaxis for mucosal bleeding, such as GI bleeding and menorrhagia, was not found to be as effective as that for joint bleeding. The rate of favorable outcomes with regular prophylaxis in GI bleeding was 49%, compared to 86% for joint bleeding.37 This lower efficacy was also demonstrated in their follow-up study of prophylaxis when a dose escalation strategy was used.38 Interestingly, in the only randomized controlled trial available to date that addresses this topic, prophylaxis resulted in more GI hemorrhage (response rate, 13.87; 95% CI, 1.84-104.46) than on-demand treatment.39 However, most of the GI bleeding events in this study occurred in a single patient, and there is no reason to suspect that result would be replicated in a larger study. Overall, further study is needed into the optimal prophylaxis regimens in GI bleeding for VWD, but the available evidence from registry studies suggests a decreased number and intensity of GI bleeding events.6,38,40,41
Adjuvant and rescue treatments
Other treatments outside the traditional hemostatic management or procedural interventions that have been used in case reports are further described in Table 2.11 A number of these adjunctive or rescue treatments, such as thalidomide or statin therapies, focus on targeting angiogenesis pathways.11 Atorvastatin is the most used and would be the recommended choice of statin were one to be used. Data are lacking as to whether other statins are less effective.
Adjunctive and rescue treatments for VWD-associated GI bleeding
| Treatment | Time course | Mechanism | Notes |
|---|---|---|---|
| High-dose statins11 | Nonacute | Antiangiogenic | |
| Bevicizumab11 | Nonacute | Decreases angiogenesis by acting as humanized monoclonal antibody against VEGF | Also used in HHT; bleeding risk |
| Octreotide11 | Acute and prophylactic management | Decreased splanchnic blood flow and improved platelet aggregation | |
| Thalidomide11,24 | Nonacute | Antiangiogenic | Based on limited data, appears less effective than lenalidomide |
| Lenalidomide42 | Nonacute, up titration as needed, potential for long-term control | Antiangiogenic | |
| Estrogen/progesterone43 | Acutely | Stimulation of ECs to increase VWF | Potential thrombotic side effects |
| Tamoxifen44 | Nonacute | Antiangiogenic | Also used in HHT |
| Steroids (including danazol)11 | Acutely | Increase in FVIII levels | Often transient effect, potentially toxic to the liver |
| β blockers (usually nonselective, such as propranolol) | Both acutely and prophylactically | Blood flow redistribution | Usually accompanied by octreotide |
| Treatment | Time course | Mechanism | Notes |
|---|---|---|---|
| High-dose statins11 | Nonacute | Antiangiogenic | |
| Bevicizumab11 | Nonacute | Decreases angiogenesis by acting as humanized monoclonal antibody against VEGF | Also used in HHT; bleeding risk |
| Octreotide11 | Acute and prophylactic management | Decreased splanchnic blood flow and improved platelet aggregation | |
| Thalidomide11,24 | Nonacute | Antiangiogenic | Based on limited data, appears less effective than lenalidomide |
| Lenalidomide42 | Nonacute, up titration as needed, potential for long-term control | Antiangiogenic | |
| Estrogen/progesterone43 | Acutely | Stimulation of ECs to increase VWF | Potential thrombotic side effects |
| Tamoxifen44 | Nonacute | Antiangiogenic | Also used in HHT |
| Steroids (including danazol)11 | Acutely | Increase in FVIII levels | Often transient effect, potentially toxic to the liver |
| β blockers (usually nonselective, such as propranolol) | Both acutely and prophylactically | Blood flow redistribution | Usually accompanied by octreotide |
HHT, hereditary hemorrhagic telangiectasia.
Other interventions, such as steroids, are thought to increase the levels of circulating VWF. The rationale behind many of these treatments is to reduce the contribution of angiodysplasia to these GI bleeds. Therapies used before these rescue treatments are initiated are highly variable. Some of these therapies require a longer time course for effectiveness and should, if needed, be used alongside, rather than in place of, previously described treatments.
Often, multiple adjunctive treatments are used together, and their variability in the time course makes it difficult to discern which intervention has had a therapeutic effect. Moreover, in more than half of all cases found in the literature on vascular abnormalities in VWD patients, the ultimate clinical outcome was not reported.11 The role of concurrent antiangiogenic therapies when combined with prophylaxis for GI bleeding warrants further study.11
Other implications for care
Overall, GI bleeding in VWD patients is difficult to manage. The existing therapies for VWD-related bleeding appear to have lower efficacy in GI bleeding. Likely, this difficulty stems from the ongoing development of angiodysplasia in VWD, which is responsible for many GI bleeds. Much further study is needed to further optimize care for these patients with regard to diagnosis, prophylaxis, and adjunctive treatments—particularly those that target angiogenesis. How many patients with angiodysplasia have undiagnosed abnormalities related to VWF also remains unknown, whether they are a result of VWD or AVWS.
CLINICAL CASE (Continued)
Over the next 2 months, despite prophylaxis, the patient has an ongoing weekly transfusion requirement and 3 readmissions for GI bleeding, with angiodysplasia treated with activated protein C for 2 of them. A decision is made prior to discharge on the third hospitalization to start the patient on atorvastatin, 80 mg daily, in an attempt to address his angiodysplasia. Over the next 3 months, his transfusion requirement is eliminated, and subsequently, the daily prophylaxis frequency is reduced. One year after starting atorvastatin, the patient has remained free from hospitalization for bleeding events for over 6 months.
Conflict-of-interest disclosure
Paula D. James: research funding: Bayer, CSL Behring, Takeda; consultancy: Band Therapeutics.
Nicholas L.J. Chornenki: no competing financial interests to declare.
Edwin Ocran: no competing financial interests to declare.
Off-label drug use
Nicholas L.J. Chornenki: tranexamic acid is discussed in Table 1; statins (atorvastatin), bevicizumab, octreotide, lenalidomide, estrogen/progesterone, tamoxifen, steroids (prednisone), danazol, and beta blockers (propranolol) are discussed in Table 2.
