


Abstract
In this review, we present a clinical case report and discussion to outline the importance of long-term specific Fanconi anemia (FA) monitoring, and we discuss the main aspects of the general management of patients with FA and clinical complications. While several nontransplant treatments are currently under evaluation, hematopoietic stem cell transplantation (HSCT) remains the only therapeutic option for bone marrow failure (BMF). Although HSCT outcomes in patients with FA have remarkably improved over the past 20 years, in addition to the mortality intrinsic to the procedure, HSCT increases the risk and accelerates the appearance of late malignancies. HSCT offers the best outcome when performed in optimal conditions (moderate cytopenia shifting to severe, prior to transfusion dependence and before clonal evolution or myelodysplasia/acute myeloid leukemia); hence, an accurate surveillance program is vital. Haploidentical HSCT offers very good outcomes, although long-term effects on malignancies have not been fully explored. A monitoring plan is also important to identify cancers, particularly head and neck carcinomas, in very early phases. Gene therapy is still experimental and offers the most encouraging results when performed in early phases of BMF by infusing high numbers of corrected cells without genotoxic effects. Patients with FA need comprehensive monitoring and care plans, coordinated by centers with expertise in FA management, that start at diagnosis and continue throughout life. Such long-term follow-up is essential to detect complications related to the disease or treatment in this setting.
Learning Objectives
Review modern management of Fanconi anemia and potential clinical complications of the disease
Emphasize importance of long-term monitoring plan of patients with FA mainly focused on malignancies
Introduction
Fanconi anemia (FA) is a rare, multisystem, genetic disorder, characterized by bone marrow failure (BMF); somatic malformations; cancer predisposition, mainly for acute myeloid leukemia (AML) and epithelial cancers of the head and neck; and exquisite sensitivity to proinflammatory cytokines and alkylating agents.1,2
FA is caused by biallelic inactivating mutations in 1 of the 23 genes of the FA/BRCA DNA repair pathway.3,4 In addition to DNA repair, FA proteins are involved in numerous functions, including aldehyde detoxification,5 inflammatory cytokine hypersensitivity,6,7 cell cycle regulation via the p53/p21 pathway,8 and oxidative phosphorylation.9,10 Disruption of these functions contributes to the exhaustion of hematopoietic stem cells (HSCs), leading to BMF. In this review, we present a case report that outlines the importance of a long-term specific monitoring plan, and we discuss the main aspects of the general management of patients with FA.
CLINICAL CASE
A newborn native of Bangladesh, born from unrelated parents, with a perinatal history of intrauterine growth retardation and small gestational age, presented at 15 days of life with duodenal stenosis requiring duodenal-jejunum surgical anastomosis. Clinical phenotype also included failure to thrive, microcephaly, triangular visage, hypotelorism, microphthalmia, and right kidney ectopia. Diepoxybutane analysis, routinely performed as a screening procedure in our institution for all newborns with multiple congenital malformations, was positive, and a homozygous mutation c.1788T>A p. Tyr596X in the FANCG gene inherited by each heterozygous parent was detected. The patient received a diagnosis of FA at 1 month of age in the absence of hematologic abnormalities and initiated the monitoring plan used in our center.1
Diagnosis, management, and follow-up postdiagnosis
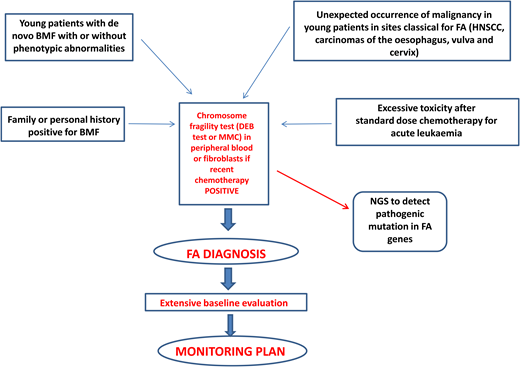
Diagnosis of FA should always be suspected in the presence of BMF with or without associated classical malformations and in young patients with de novo BMF, positive family history for BMF, spontaneous chromosomal breaks, or unbalanced 1q, 3q, or 7q translocations found during the diagnostic workup for myelodysplasia (MDS) or acute myeloid leukemia (AML).1 Furthermore, the presence of early-onset tumors in sites classical for FA and excessive toxicity after standard-dose chemotherapy should also trigger suspicion of an underlying FA diagnosis.1 Diagnostic confirmation with the chromosomal fragility test after exposure to DNA cross-linking agents (diepoxybutane or mitomycin C) is mandatory since this analysis still represents the gold standard for the diagnosis of FA. Mosaicism can occur as a consequence of molecular events (back mutation, intragenic crossover, gene conversion, compensating deletions/insertion, or gain-of-function mutations), generating a correction of a FA mutation in a hematopoietic stem cell or in a lymphocyte progenitor. In this case, the chromosomal fragility test on peripheral blood may be negative, but if the suspicion of FA persists, the test should be performed on “nonblood” chromosomes (skin fibroblasts or hair follicles). If positive, it leads to the identification of a mosaic FA.1
Next-generation sequencing has proven to be efficient and accurate in the diagnosis of FA and other inherited marrow failures11 and is now part of the routine diagnostic workup. The next-generation sequencing approach has some difficulties related to the interpretation or even to the inconclusiveness of results, which may require more comprehensive tools such as whole- exome or whole-genome sequencing.1,12 Complementation analysis may still be valuable as a postmolecular functional confirmatory test in dubious cases.
After diagnosis, patients with FA should undergo baseline extensive multiorgan assessment (Table 1) prior to enter a monitoring plan designed with follow-up by a hematology oncology center specializing in inherited bone marrow failure syndromes.1 The goal for the BMF monitoring plan is to intercept the best time for hematopoietic stem cell transplantation (HSCT) and to evaluate the degree of cytopenia, the presence of marrow dysplastic features or MDS or AML, the presence and type of cytogenetic abnormalities, and infections, transfusion loads, types of available donors, availability of stem cells, and efficacy of nontransplant treatment1,13 (Figure 1).
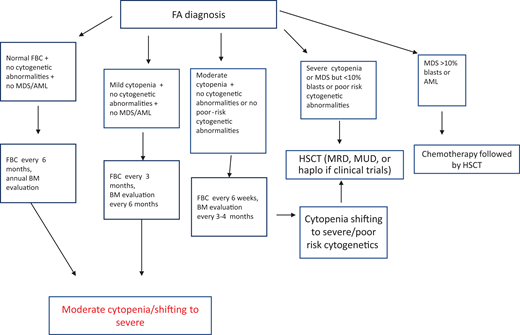
Postdiagnosis hematologic monitoring and decision-making process. Modified from Dufour.1 BM, bone marrow; FBC, full blood count; haplo, haploidentical donor; MRD, matched related donor; MUD, matched unrelated donor.
Postdiagnosis hematologic monitoring and decision-making process. Modified from Dufour.1 BM, bone marrow; FBC, full blood count; haplo, haploidentical donor; MRD, matched related donor; MUD, matched unrelated donor.
Workup for patients with FA after diagnosis
| Soon after diagnosis: |
|---|
| Hematologic evaluation • FBC with differential count • Serum immunoglobulin levels, peripheral blood immunophenotyping, response to vaccines • Bone marrow aspiration for morphology and cytogenetics and immunophenotyping • Trephine bone marrow biopsy (definition of hematopoietic cellularity, abnormal localization of myeloid progenitors, increased blasts on specific CD34 and CD117 staining) |
| Extensive extra-hematologic evaluation • Liver, kidney, heart, urinary tract, gastrointestinal tract, skeleton, hearing and visual function evaluation • Endocrinologic evaluation (thyroid, glucose tolerance, pituitary gland function, gonads in postpubertal subjects) • Psychological evaluation • Brain magnetic resonance imaging with angiography for pituitary gland and Moyamoya syndrome • Assessment for cancers typically associated to FA with special attention to oral cavity |
| Chromosomal fragility tests in siblings and possibly in relatives |
| HLA typing of the patient, healthy sibling, and parents |
| Search of HLA-matched unrelated donor if no healthy HLA-matched sibling is found in the family |
| Modified from Dufour.1 |
| FBC, full blood count; HLA, human leukocyte antigen. |
| Soon after diagnosis: |
|---|
| Hematologic evaluation • FBC with differential count • Serum immunoglobulin levels, peripheral blood immunophenotyping, response to vaccines • Bone marrow aspiration for morphology and cytogenetics and immunophenotyping • Trephine bone marrow biopsy (definition of hematopoietic cellularity, abnormal localization of myeloid progenitors, increased blasts on specific CD34 and CD117 staining) |
| Extensive extra-hematologic evaluation • Liver, kidney, heart, urinary tract, gastrointestinal tract, skeleton, hearing and visual function evaluation • Endocrinologic evaluation (thyroid, glucose tolerance, pituitary gland function, gonads in postpubertal subjects) • Psychological evaluation • Brain magnetic resonance imaging with angiography for pituitary gland and Moyamoya syndrome • Assessment for cancers typically associated to FA with special attention to oral cavity |
| Chromosomal fragility tests in siblings and possibly in relatives |
| HLA typing of the patient, healthy sibling, and parents |
| Search of HLA-matched unrelated donor if no healthy HLA-matched sibling is found in the family |
| Modified from Dufour.1 |
| FBC, full blood count; HLA, human leukocyte antigen. |
Regarding the risk of head and neck squamous cell carcinomas, patients should be encouraged to avoid alcohol and tobacco and to maintain good oral hygiene. The oral cavity should be examined every 6 months by an expert in oral cancers. A large German study14 showed that brushing of visible lesions and cytology analysis are highly sensitive (97.7%) and specific (84.5%) to identify premalignant and malignant lesions. The addition of DNA ploidy analysis to cytologic examination increased the sensitivity and specificity to 100% and 92.2%, respectively. Importantly, 63% of malignant lesions were detected at an early stage and were curable only with surgery, indicating that this is an effective tool for early diagnosis of cancers of the oral cavity.14
Patients with mutations in FANCD1/BRCA2 or FANCN/PALB2 need specific cancer screenings (ie, brain magnetic resonance imaging and kidney ultrasound) because of increased risk of medulloblastoma, Wilms tumor, and other cancers.1
In Table 2, we summarize the cancer screening guidelines of the American Association of Cancer Research and Fanconi Anemia Research Fund.15,16
Long-term follow-up of extra-hematologic manifestations in transplants related to underlying disease in patients with FA
| Organ or system affected | Adverse effects | Causes | Follow-up |
|---|---|---|---|
| General | Short stature Primary or secondary cancer | FA, HSCT FA, HSCT, cGVHD | Annual Dermatology skin cancer screening every 6-12 months Oral self-examinations (or with parents' assistance) once a month Oral cavity examination every 6 months Annual HNSCC examination by an otolaryngologist beginning in early adolescence Annual gynecologic evaluation MRI to detect breast cancer from age 20 Annual esophagoscopy/coloscopy from age 20 Abdominal ultrasound annually to detect liver/kidney tumors or neuroblastoma Increased screening for patients with cGVHD and BRCA2 mutation |
| Skin | Pigmentation, dryness, thickening | FA, cGVHD | Annual |
| CNS | Side effects of radiation Moyamoya syndrome Congenital malformations | HSCT FA | Annual |
| Eyes | Cataract, retinitis Very dry eyes (sicca syndrome) | HSCT cGVHD | Annual As needed |
| Ear, nose, and throat | Sinusitis, very dry mouth (sicca syndrome) Hearing loss | cGVHD FA HSCT | As needed Annual |
| Heart | Congenital anomalies Iron overload | FA Transfusions | As needed As needed |
| Lungs | Side effects of HSCT | HSCT, GVHD | As needed |
| Liver | Chronic liver disease (transaminitis) or cholestasis Iron overload | HSCT, GVHD Transfusions | As needed As needed |
| Kidneys and genitourinary system | Congenital anomalies Chronic renal insufficiency | FA HSCT | As needed As needed |
| GI tract | Congenital anomalies Failure to thrive, functional problems (eg, malabsorption) | FA FA, GVHD | As needed As needed |
| Endocrine | Diabetes Hypothyroidism | FA, GVHD (steroids) FA, HSCT | Annual, if present as needed Annual |
| Gonadal | Virilization Infertility, early menopause | Androgen treatment FA, HSCT | As needed Annual |
| Musculoskeletal | Hand and arm abnormalities Hip dysplasia | FA FA | As needed As needed |
| Psychological | Psychosocial issues (eg, anxiety, depression) | FA, GVHD, HSCT | As needed |
| Modified from Fanconi Anemia Research Fund guidelines16 and Walsh et al.15 | |||
| CNS, central nervous system; GI, gastrointestinal; HNSCC, head and neck squamous cell carcinoma; MRI, magnetic resonance imaging. | |||
| Organ or system affected | Adverse effects | Causes | Follow-up |
|---|---|---|---|
| General | Short stature Primary or secondary cancer | FA, HSCT FA, HSCT, cGVHD | Annual Dermatology skin cancer screening every 6-12 months Oral self-examinations (or with parents' assistance) once a month Oral cavity examination every 6 months Annual HNSCC examination by an otolaryngologist beginning in early adolescence Annual gynecologic evaluation MRI to detect breast cancer from age 20 Annual esophagoscopy/coloscopy from age 20 Abdominal ultrasound annually to detect liver/kidney tumors or neuroblastoma Increased screening for patients with cGVHD and BRCA2 mutation |
| Skin | Pigmentation, dryness, thickening | FA, cGVHD | Annual |
| CNS | Side effects of radiation Moyamoya syndrome Congenital malformations | HSCT FA | Annual |
| Eyes | Cataract, retinitis Very dry eyes (sicca syndrome) | HSCT cGVHD | Annual As needed |
| Ear, nose, and throat | Sinusitis, very dry mouth (sicca syndrome) Hearing loss | cGVHD FA HSCT | As needed Annual |
| Heart | Congenital anomalies Iron overload | FA Transfusions | As needed As needed |
| Lungs | Side effects of HSCT | HSCT, GVHD | As needed |
| Liver | Chronic liver disease (transaminitis) or cholestasis Iron overload | HSCT, GVHD Transfusions | As needed As needed |
| Kidneys and genitourinary system | Congenital anomalies Chronic renal insufficiency | FA HSCT | As needed As needed |
| GI tract | Congenital anomalies Failure to thrive, functional problems (eg, malabsorption) | FA FA, GVHD | As needed As needed |
| Endocrine | Diabetes Hypothyroidism | FA, GVHD (steroids) FA, HSCT | Annual, if present as needed Annual |
| Gonadal | Virilization Infertility, early menopause | Androgen treatment FA, HSCT | As needed Annual |
| Musculoskeletal | Hand and arm abnormalities Hip dysplasia | FA FA | As needed As needed |
| Psychological | Psychosocial issues (eg, anxiety, depression) | FA, GVHD, HSCT | As needed |
| Modified from Fanconi Anemia Research Fund guidelines16 and Walsh et al.15 | |||
| CNS, central nervous system; GI, gastrointestinal; HNSCC, head and neck squamous cell carcinoma; MRI, magnetic resonance imaging. | |||
Clonal evolution
Patients with FA have a 30% to 40% cumulative risk of myeloid malignancies by age 40 years.17,18 These often arise from clonal karyotype abnormalities and unbalanced translocations, leading to gains or losses of chromosomes. Some cytogenetic changes predict negative outcomes, and their identification is important to decide the timing of HSCT19,20 (Figure 1).
Gain of chromosome 1q is the most common abnormality in FA; it can occur without MDS/AML and does not necessarily indicate impending transformation.18 Conversely, monosomy 7/del(7q) is considered a marker of transformation, requiring urgent HSCT.18 Other cytogenetic changes indicating a poor prognosis are gain of chromosome 3q, frequently preceding monosomy 7/del(7q),21,22 and RUNX1 abnormalities, including cryptic translocations, that also indicate immediate HSCT.23
An urgent HSCT is also needed in case of overt MDS/AML, and the most attention should be paid to prevent this condition since it has been shown that transplants performed with active MDS/AML have a significantly lower overall survival (OS) vs those performed in complete remission.24 These findings reinforce the need for an accurate monitoring plan to intercept the “momentum” and suggest that a cytoreductive approach, withstanding the risks of undesired toxicity, should not be avoided because of the fear of negative toxic effects during post-HSCT.
Regarding pre-HSCT chemotherapy, the FLAG regimen (fludarabine 30 mg/m² daily, cytarabine 1 g/m² twice daily for five days plus G-CSF), followed by HSCT in the aplastic phase (without waiting for hematopoietic recovery), described in a French-Brazilian study, led to a 3-year progression-free survival and OS of 53% and 53%, respectively, although there was a high rate of fungal and viral infections.25
CLINICAL CASE (Continued)
At the age of 3 years, the patient developed a mild cytopenia (neutrophils 1.1 × 109/L and platelets 100 × 109/L). Bone marrow assessment showed cellularity of 70% (trephine biopsy), no dysplastic signs, and normal karyotype.
According to our institutional protocol1 (Figure 1), he received complete blood counts every 3 months and bone marrow evaluation every 6 months. Both remained stable over the following 2 years.
At the age of 6 years, platelets declined to 70 × 109/L. At the next control 3 months later, they suddenly dropped to 7 × 109/L. At this point in time, marrow cytogenetic analysis showed a complex abnormality (46, XY, add(21)(q22)7 /46,XY[]5) and a RUNX1 breakage in 34 of 100 nuclei analyzed by fluorescence in situ hybridization.
This monitoring program enabled the identification of an unexpected condition that required urgent HSCT. No human leukocyte antigen–matched family donor was available, and the poor prognosis cytogenetic anomalies created a major timing concern in identifying a suitable matched unrelated donor.
HSCT indications
HSCT is the only cure for BMF, but in addition to the mortality intrinsic to the procedure, it is also known to increase the risk and accelerate the appearance of late malignancies.26,27 Therefore, indications for transplant should be carefully weighed, based on a thorough evaluation of risks and benefits.
Established indications for HSCT in FA include severe cytopenia or progression of moderate cytopenia, poor prognosis cytogenetic aberrations, and overt MDS/AML.1,13,28
Special attention is warranted for BRCA2 patients, who, due to the extremely high occurrence of AML (cumulative incidence [CI] of 80%) and solid cancers (CI of 97%) within the first 10 years of age,29 may be considered for preemptive HSCT. The optimal time for HSCT is when cytopenia shifts from moderate to severe, prior to transfusion dependency, probably before androgen therapy, and before clonal evolution.1,13
Over the past 20 years, the outcome of HSCT in patients with FA has improved dramatically because of reduction of the doses of alkylating agents and irradiation, the introduction of fludarabine, and the use of recipient T-cell depletion (TCD) that improved engraftment and graft-vs-host disease (GVHD) rates.30 In the different donor settings, OS peaks from 84% to 95% and above, with variable rates of post-HSCT malignancies.
Tables 3 to 5 summarize the outcomes of the largest cohort of patients with FA who underwent transplantation from matched related donor (MRD), matched unrelated donor (MUD), and alternative donor (AD).
Outcome of the largest cohort of patients with FA who underwent transplantation from a matched related donor
| Reference | Patients (n) | Prevalently using conditioning | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Malignancies, No. (%) |
|---|---|---|---|---|---|---|---|---|
| Pasquini et al45 (2008) | 148 | Cy ± ATG ± Bus TAI, TLI, TBI + Cy ± ATG | 5 years (censored) | 19% (grades II-IV) | 20% at 5 years | 17.5% (overall) | 78% (IRR) 81% (no IRR) | 4/148 (2.7%) |
| Peffault de Latour et al26 (2013) | 211 (post 1999) | Flu + ATG ± IRR | 6 years | 36% (grades III-IV) | 20% at 5 years | 14% | 76% at 5 years | 15-year CI 15% overall |
| Benajiba et al46 (2015) | 20 | Flu + Cy | 2 years | 15% (grades III-IV) | 10% extensive 15% limited | 5% | 95% at 2 years | 0 |
| Bonfim et al47 (2019) | 91 | Cy + ATG | 3-7 years | 11% (grades II-IIV) | 23.5% | 7% | 95% at 5 years | 1/43 (2.3%) |
| Bernard et al48 (2021) | 42 | Flu-Cy-alemtuzumab | 74.4 months | 6.1% (grades II-IV) overall | 2.4% overall | 13.8% overall | 85.4% at 5 years | 3/82 (3.6%) overall |
| Modified from Pierri et al.13 | ||||||||
| ATG, antithymocyte globulin; Bus, busulfan; Cy, cyclophosphamide; Flu, fludarabine; IRR, irradiation in conditioning regimen; NR, not reported; TAI, thoraco-abdominal irradiation; TBI, total body irradiation; TLI, total lymph node irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality. | ||||||||
| Reference | Patients (n) | Prevalently using conditioning | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Malignancies, No. (%) |
|---|---|---|---|---|---|---|---|---|
| Pasquini et al45 (2008) | 148 | Cy ± ATG ± Bus TAI, TLI, TBI + Cy ± ATG | 5 years (censored) | 19% (grades II-IV) | 20% at 5 years | 17.5% (overall) | 78% (IRR) 81% (no IRR) | 4/148 (2.7%) |
| Peffault de Latour et al26 (2013) | 211 (post 1999) | Flu + ATG ± IRR | 6 years | 36% (grades III-IV) | 20% at 5 years | 14% | 76% at 5 years | 15-year CI 15% overall |
| Benajiba et al46 (2015) | 20 | Flu + Cy | 2 years | 15% (grades III-IV) | 10% extensive 15% limited | 5% | 95% at 2 years | 0 |
| Bonfim et al47 (2019) | 91 | Cy + ATG | 3-7 years | 11% (grades II-IIV) | 23.5% | 7% | 95% at 5 years | 1/43 (2.3%) |
| Bernard et al48 (2021) | 42 | Flu-Cy-alemtuzumab | 74.4 months | 6.1% (grades II-IV) overall | 2.4% overall | 13.8% overall | 85.4% at 5 years | 3/82 (3.6%) overall |
| Modified from Pierri et al.13 | ||||||||
| ATG, antithymocyte globulin; Bus, busulfan; Cy, cyclophosphamide; Flu, fludarabine; IRR, irradiation in conditioning regimen; NR, not reported; TAI, thoraco-abdominal irradiation; TBI, total body irradiation; TLI, total lymph node irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality. | ||||||||
Outcome of the largest cohort of patients with FA who underwent transplantation from matched unrelated donors
| Reference | Patients (n) | Conditioning regimens | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Secondary malignancies |
|---|---|---|---|---|---|---|---|---|
| Wagner et al49 (2007) | 98 | No Flu (Cy ± Bus ± ATG + IRR) Yes Flu (Flu + Cy ± ATG ± IRR ± Bus) | 3 years | 16% Flu + TCD 70% No Flu No TCD | 31% | 47% Flu 81% no Flu at 3 years | 52% Flu 13% no Flu at 3 years | NR |
| Peffault de Latour et al26 (2013) | 176 post-1999 | Flu ATG ± IRR | 6 years | 36% (grades III-IV) | 16% at 5 years | 27% at 5 years | 64% at 5 years | 15 years CI 15% overall |
| Bernard et al48 (2021) | 23 | Flu-Cy-alemtuzumab or ATG | 74.4 months | 6.1% overall | 2.4% overall | 13.8% overall | 95.7% at 5 years | NR |
| Bonfim et al34 (2022) | 51 | Flu-Cy-ATG | 5 years | 16% | 27% | NR | 83% | NR |
| Modified from Pierri et al.13 | ||||||||
| ATG, antithymocyte globulin; Bus, busulfan; Cy, cyclophosphamide; Flu, fludarabine; IRR, irradiation in conditioning regimen; NR, not reported; TAI, thoraco-abdominal irradiation; TBI, total body irradiation; TLI, total lymph node irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality. | ||||||||
| Reference | Patients (n) | Conditioning regimens | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Secondary malignancies |
|---|---|---|---|---|---|---|---|---|
| Wagner et al49 (2007) | 98 | No Flu (Cy ± Bus ± ATG + IRR) Yes Flu (Flu + Cy ± ATG ± IRR ± Bus) | 3 years | 16% Flu + TCD 70% No Flu No TCD | 31% | 47% Flu 81% no Flu at 3 years | 52% Flu 13% no Flu at 3 years | NR |
| Peffault de Latour et al26 (2013) | 176 post-1999 | Flu ATG ± IRR | 6 years | 36% (grades III-IV) | 16% at 5 years | 27% at 5 years | 64% at 5 years | 15 years CI 15% overall |
| Bernard et al48 (2021) | 23 | Flu-Cy-alemtuzumab or ATG | 74.4 months | 6.1% overall | 2.4% overall | 13.8% overall | 95.7% at 5 years | NR |
| Bonfim et al34 (2022) | 51 | Flu-Cy-ATG | 5 years | 16% | 27% | NR | 83% | NR |
| Modified from Pierri et al.13 | ||||||||
| ATG, antithymocyte globulin; Bus, busulfan; Cy, cyclophosphamide; Flu, fludarabine; IRR, irradiation in conditioning regimen; NR, not reported; TAI, thoraco-abdominal irradiation; TBI, total body irradiation; TLI, total lymph node irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality. | ||||||||
Outcome of the largest cohort of patients with FA who underwent transplantation from alternative donors
| Reference | Patients (n) | Donor type | Prevalently using conditioning | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Malignancies, No. (%) |
|---|---|---|---|---|---|---|---|---|---|
| MacMillan et al50 (2015) | 130 | 73 MUD (TCD) 39 1-2 MM (no TCD) CB 18 1Ag MM Unrelated (TCD) | Flu + Cy + ATG + TBI (in the most recent patients) with thymic shielding | 7.7 years | 20% (grades II-IV) | 10% | NR | 58% at 5 years | 11/130 (8.4%) |
| Mehta et al51 (2017) | 45 | 25 MUD 14 MMUD 6 MMRD All TCD | Bu-Flu-Cy-ATG | 41 months | 6.7% (0 grades III-IV) | 3/45 (6.7%) limited, 0 extensive | NR | 80% at 3 years | 1/45 (2.2%) |
| Ebens et al52 (2018) | 57 | All TCD 32 (m)MUD BM 20 (m)MUD UCB 2 MMRD 1 8/8 HLA parental BM 1 MMSD UCB + BM boost | Flu-Cy-TBI-ATG | 6.2 years | 12% grades II-IV | 4/57 (7%) | NR | 86% at 5 years | NR |
| Zubicaray et al35 (2021) | 123 | MMUD (all TCD) | Flu-Cy-TBI-ATG | 44.8 months | 41% grades II-IV | 5% limited 10% extensive | NR | 62% at 2 years | NR |
| Modified from Pierri et al.13 | |||||||||
| ATG, antithymocyte globulin; BM, bone marrow; Bus, busulfan; CB, cord blood; Cy, cyclophosphamide; Flu, fludarabine; HLA, human leukocyte antigen; IRR, irradiation in conditioning regimen; MM, mismatched; MMRD, mismatched related donor; MMUD, mismatched unrelated donor; MUD, matched unrelated donor; NR, not reported; TBI, total body irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality; UCB, unrelated cord blood. | |||||||||
| Reference | Patients (n) | Donor type | Prevalently using conditioning | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Malignancies, No. (%) |
|---|---|---|---|---|---|---|---|---|---|
| MacMillan et al50 (2015) | 130 | 73 MUD (TCD) 39 1-2 MM (no TCD) CB 18 1Ag MM Unrelated (TCD) | Flu + Cy + ATG + TBI (in the most recent patients) with thymic shielding | 7.7 years | 20% (grades II-IV) | 10% | NR | 58% at 5 years | 11/130 (8.4%) |
| Mehta et al51 (2017) | 45 | 25 MUD 14 MMUD 6 MMRD All TCD | Bu-Flu-Cy-ATG | 41 months | 6.7% (0 grades III-IV) | 3/45 (6.7%) limited, 0 extensive | NR | 80% at 3 years | 1/45 (2.2%) |
| Ebens et al52 (2018) | 57 | All TCD 32 (m)MUD BM 20 (m)MUD UCB 2 MMRD 1 8/8 HLA parental BM 1 MMSD UCB + BM boost | Flu-Cy-TBI-ATG | 6.2 years | 12% grades II-IV | 4/57 (7%) | NR | 86% at 5 years | NR |
| Zubicaray et al35 (2021) | 123 | MMUD (all TCD) | Flu-Cy-TBI-ATG | 44.8 months | 41% grades II-IV | 5% limited 10% extensive | NR | 62% at 2 years | NR |
| Modified from Pierri et al.13 | |||||||||
| ATG, antithymocyte globulin; BM, bone marrow; Bus, busulfan; CB, cord blood; Cy, cyclophosphamide; Flu, fludarabine; HLA, human leukocyte antigen; IRR, irradiation in conditioning regimen; MM, mismatched; MMRD, mismatched related donor; MMUD, mismatched unrelated donor; MUD, matched unrelated donor; NR, not reported; TBI, total body irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality; UCB, unrelated cord blood. | |||||||||
CLINICAL CASE (Continued)
Due to time constraints in relation to severe cytopenia and cytogenetic abnormalities, the patient underwent HSCT from his haploidentical mother with the TCD platform in use in our center (α/β/CD19 depletion) at the age of 6 years.
Conditioning regimen included fludarabine 120 mg/m2, cyclophosphamide 1200 mg/m2, 200 cGy single-dose total body irradiation pretransplant antithymocyte globulin (ATG) (Grafalon; Neovii Biotech) 4 mg/kg/d for 3 days, and rituximab (200 mg/m2) on day −1.
The patient received >8 × 106/kg CD34 cells/kg, <1 × 105/kg T cell receptor (TCR) αβ, and <2 × 105/kg CD19 cells according to the protocol in use at our center.31 No posttransplantation pharmacologic immunosuppression was administered.
The posttransplant course was uncomplicated without acute GHVD (aGVHD) and chronic GVHD (cGVHD). Presently, at the age of 11 years, at 5 years post-HSCT, the boy is well (Karnofsky score of 100%) and under a surveillance plan, without complications related to HSCT or the underlying disease.
Haploidentical HSCT in FA
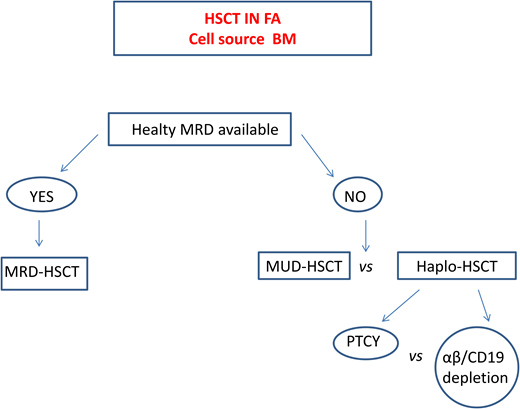
In patients lacking a standard donor, haploidentical HSCT is a promising strategy. This procedure offers virtually all patients the chance of an HSCT with a fast accessibility. The use of megadoses of HSCs and the depletion from the graft of alloreactive donor T lymphocytes13,32 significantly reduced the incidence of graft rejection and of GVHD initially observed. Table 6 summarizes the outcomes of the largest cohorts of patients with FA who underwent transplantation from haploidentical donors.
Outcome of the largest cohort of patients with FA who underwent transplantation from haploidentical donors
| Reference | Patients (n) | Prevalently using conditioning regimen | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Malignancies, % |
|---|---|---|---|---|---|---|---|---|
| Bonfim et al53 (2017) | 26 | Flu-mini TBI ± ATG + PTCY | 30 months | 75% no ATG 14.2% ATG | 83% no ATG 14.2% ATG | 41.6% no ATG 21.4% ATG | 72.6% at 1 year | NR |
| Ayas et al54 (2019) | 19 | Flu-mini TBI-ATG + PTCY | 38.3 + 5.8 months | Grades III-IV 15.7% | Extensive in 1 patient (5.3%) | 5.2% | 89.2% + 7.2% at 5 years | None |
| Bonfim et al34 (2022) | 49 | Flu-mini TBI-ATG or alemtuzumab | 3.8 years | Grades II-IV 28% | 26% | 8/49 (16.3%) | 82% at 2 years | 4% |
| Strocchio et al33 (2021) | 24 | Flu-CY-mini TBI Alfa/beta/CD19 depletion | 5.2 years | Grades I-II 17.4% | 5.5% | 0 | 100% at 5 years | 4.1% |
| Reference | Patients (n) | Prevalently using conditioning regimen | Follow-up | aGVHD | cGVHD | TRM/NRM | OS | Malignancies, % |
|---|---|---|---|---|---|---|---|---|
| Bonfim et al53 (2017) | 26 | Flu-mini TBI ± ATG + PTCY | 30 months | 75% no ATG 14.2% ATG | 83% no ATG 14.2% ATG | 41.6% no ATG 21.4% ATG | 72.6% at 1 year | NR |
| Ayas et al54 (2019) | 19 | Flu-mini TBI-ATG + PTCY | 38.3 + 5.8 months | Grades III-IV 15.7% | Extensive in 1 patient (5.3%) | 5.2% | 89.2% + 7.2% at 5 years | None |
| Bonfim et al34 (2022) | 49 | Flu-mini TBI-ATG or alemtuzumab | 3.8 years | Grades II-IV 28% | 26% | 8/49 (16.3%) | 82% at 2 years | 4% |
| Strocchio et al33 (2021) | 24 | Flu-CY-mini TBI Alfa/beta/CD19 depletion | 5.2 years | Grades I-II 17.4% | 5.5% | 0 | 100% at 5 years | 4.1% |
ATG, antithymocyte globulin; Cy, cyclophosphamide; Flu, fludarabine; NR, not reported; PTCY, posttransplantation cyclophosphamide; TBI, total body irradiation; TRM/NRM, transplant-related mortality/nonrelapse mortality.
Currently, 2 platforms for TCD are used. In one, the TCR αβ+ cells are depleted via graft manipulation, whereas in the other, specific T-cell subpopulations (e.g., TCR γδ+) that have graft protective and graft-vs-leukemia (GVL) effects are retained.32 A cohort of 24 patients with FA who underwent transplantation with this platform showed a 2-year event-free survival (EFS) of 86.3% and grade I to II aGVHD and cGVHD of 17.4% and 5.5%, respectively.33 However, since this TCD system is expensive, technically demanding, and inaccessible in many countries, other groups prefer to use strategies based on posttransplant cyclophosphamide (PTCY) at a reduced dose to limit the severity of mucositis. In a Brazilian study, a subgroup of patients in whom serotherapy (ATG or alemtuzumab) was added to this platform displayed an OS of 82% but acute grade II to IV and moderate to severe cGVHD rates of 28% and 26%, respectively.34 Delayed immune reconstitution and viral reactivation remain major concerns in this setting.
A recent comparative European Society for Blood and Marrow Transplantation study35 reported superior 2-year OS (80%) and EFS (86%) in patients receiving haploidentical HSCT with in vivo TCD compared with those who received haplo transplant with ex vivo TCD (2-year OS 60%, EFS 56%), who in turn had a far lower grade II aGVHD (17%) compared with those with in vivo TCD (40%).
With the aim to ensure engraftment and improve immune reconstitution in patients with FA who underwent transplantation from an haploidentical donor, an ongoing clinical trial (NCT04784052) is evaluating a radiation-free, reduced-intensity preparative regimen containing JSP191 (ahumanized monoclonal antibody that targets CD117 and causes stem cell death). This is in combination with rabbit anti-thymocyte globuline, cyclophosphamide, fludarabine, and rituximab, followed by an infusion of αβ+ TCD donor stem cells.
Nontransplant therapy
The synthetic androgens oxymetholone and danazol were shown to produce a transient hematologic response in two-thirds of patients, rendering this treatment a reasonable option in patients who cannot access HSCT at a given time, providing them a bridge until the possibility of a transplant occurs.36
Metformin, a drug approved by the US Food and Drug Administration for treatment of diabetes mellitus, generated a hematologic response in 4 of 13 evaluable patients in a median time of 84.5 days in a phase 1 clinical trial.37
The effect on hematopoiesis of quercetin, a flavonoid polyphenol with antioxidant properties and cellular effects, including transforming growth factor β inhibition, is under evaluation in an ongoing phase 1 trial by a Cincinnati group (clinicaltrials.gov NCT01720147) in patients with FA. A preliminary report of this trial described 12 individuals treated with oral quercetin for 16 weeks, with excellent tolerance and compliance and a definite trend toward peripheral blood reactive oxygen species reduction.38 An ongoing phase 2 trial (NCT03476330) is evaluating the efficacy of a maximum daily dose of quercetin (4000 mg/d) in reducing buccal micronuclei, a surrogate marker of DNA damage with susceptibility to squamous cell carcinoma in patients with FA post-HSCT.
The thrombopoietin mimetic small molecule eltrombopag recently proved to be effective in restoring trilineage hematopoiesis in acquired severe aplastic anemia.39,40 Eltrombopag improves marrow function in severe aplastic anemia through a different mechanism. In addition to fundamental stimulation of HSCs, eltrombopag has important immunomodulatory properties (decreases interferon γ, tumor necrosis factor α, and transforming growth factor β effects), and it promotes tolerance (by increasing regulatory B and T cells) with a parallel impairment of dendritic cell maturation and macrophage activity. Furthermore, eltrombopag favors cellular iron mobilization, decreasing iron overload with a potential benefit on hematopoiesis.39 Due to its ability to improve repair of double-strand DNA breaks,41 it is now under evaluation in a prospective phase 1/2 clinical trial (NCT03204188) in patients with FA. Preliminary results show that the 4 evaluable patients who have reached the 6-month assessment end point displayed marrow response, and 2 of them displayed mono- and bilineage peripheral blood responses42 without serious adverse events.
Gene therapy in FA
At present, gene therapy (GT) in FA is available only for the FA-A genotype. Collection and correction of a sufficient number of HSPCs from patients with FA are the major obstacles to the efficacy of GT in this disease.43
In a recent update of the largest phase 1/2 Spanish long-term follow-up lentivirus clinical trial,44 the 4 of 8 evaluable patients who were infused with low numbers of corrected cells or in advanced BMF showed BMF progression requiring alternative treatment. However, 2 patients receiving higher doses of corrected CD34+ cells improved the peripheral blood cell counts. No genotoxic effect has appeared so far after GT.
Currently, GT in FA is an experimental treatment that offers the best results if performed in the early stage of BMF and when significant numbers of corrected CD34+ cells are infused. Trials using gene editing techniques are likely to be performed in the future.
Conclusion and future directions
FA is a very heterogeneous disorder with the main morbidities of BMF and increased cancer risk. No definitive treatment is available so far. HSCT is the only option currently capable of curing BMF, although it increases the risk of late malignancies already inherent in the disease. HSCT outcomes have remarkably improved over the past 20 years, thanks to the strategies aimed at reducing complications, mainly GVHD, a major determinant of posttransplant tumors. In this respect, the advent of haploidentical HSCT, with in vivo and ex vivo strategies for TCD, offers a higher accessibility to this treatment and encouraging outcomes.
HSCT provides the best results when performed in optimal conditions and intercepted by a tailored lifetime monitoring plan starting at diagnosis and performed in centers specializing in BMF disorders.
A monitoring plan should also be applied to prevent the evolution of solid tumors, mainly epithelial cancers of the head and neck and genital areas, that currently have no satisfactory treatment.
Progress in the understanding of the biology of the disease will help to develop new therapeutic strategies aimed to counteract the numerous dysregulated functions of FA cells.
Conflicts-of-interest disclosures
Carlo Dufour: Advisory Board for Novartis.
Filomena Pierri: no competing financial interests to declare.
Off-label drug use
Carlo Dufour: nothing to disclose.
Filomena Pierri: nothing to disclose.
