

Abstract
Follicular lymphoma (FL) is a heterogeneous disease, both clinically and biologically. The biological behavior and development of FL is a culmination of complex multistep processes underpinned by genetic and nongenetic determinants. Epigenetic deregulation through recurrent genetic alterations is now a recognized major biological hallmark of FL, alongside the t(14;18) translocation. In parallel, there is a strong interplay between the lymphoma B cells and the immune microenvironment, with the microenvironment serving as a critical enabler by creating a tumor-supportive niche and modulating the immune response to favor survival of the malignant B cells. A further layer of complexity arises from the biological heterogeneity that occurs between patients and within an individual, both over the course of the disease and at different sites of disease involvement. Altogether, taking the first steps to bridge the understanding of these various biological components and how to evaluate these clinically may aid and inform future strategies, including logical therapeutic interventions, risk stratification, therapy selection, and disease monitoring.
Learning Objectives
Gain an up-to-date overview of how the biology, the heterogeneity, and the evolution shape the pathogenesis of follicular lymphoma
Recognize potential opportunities in which the biology could guide and translate into the clinical arena
CLINICAL CASE
The patient is a 68-year-old woman with a 15-year history of follicular lymphoma (FL). She initially presented with a palpable left inguinal mass. The biopsy showed t(14;18)-positive grade 2 FL. Although the patient had stage 4 disease with a Follicular Lymphoma International Prognostic Index (FLIPI) score of 2, she was asymptomatic and therefore was actively observed. Four years later, she developed progressive lymphadenopathy with B symptoms. A repeat biopsy showed no evidence of transformation, and the patient was treated with immunochemotherapy (rituximab, cyclophosphamide, vincristine, and prednisolone), achieving a complete response. Approximately 5 years later, the patient developed new debilitating fatigue and progressive lymphadenopathy and was subsequently treated with rituximab and bendamustine followed by rituximab maintenance. Unfortunately, 11 months following completion of maintenance, her symptoms returned, including increased weight loss with a workup revealing progressive disease yet again. A repeat biopsy was consistent with relapsed FL. She was enrolled in a study evaluating the EZH2 inhibitor tazemetostat, and sequencing confirmed the presence of an EZH2 mutation. She achieved a partial response and continues on therapy.
Introduction
FL is the most common indolent lymphoma in Western countries, accounting for 20% of all non-Hodgkin lymphomas.1 With advances in treatment, most patients live with FL for many years, as the median survival now spans nearly 2 decades. A patient's journey is typified by a protracted relapsing and remitting pattern, whereby subsequent disease-free periods after each therapy become progressively shorter. Yet this pattern does not reflect the journey of all patients, given the recognized spectrum in clinical phenotypes. Certain patient subsets remain underserved by our current therapies. These include patients who experience multiple relapses and exhaust the majority of existing treatment modalities and those with high-risk clinical behavior who progress early after initial chemo-immunotherapy (progression of disease within 24 months) or transform to a more aggressive high-grade lymphoma, usually diffuse large B-cell lymphoma (DLBCL).2,3 These patients have significantly inferior outcomes and a higher risk of death as a result of their lymphoma.4 Elucidating the biological processes that underpin the clinical heterogeneity of FL remains a major research focus.
Major advances in our understanding of the pathogenesis of FL have been made in the last decade, with this progress made possible through advancements in high-resolution technologies.5 Over 30 years ago, the reciprocal chromosomal translocation t(14;18) was identified, and presently, there is an almost complete catalog of the genetic landscape of this lymphoma subtype. In parallel, there is accumulating evidence for the contribution of nongenomic determinants, such as the tumor microenvironment (TME) and epigenetic (de-)regulation. In addition to classical FL, a number of related histological entities have recently been described in the World Health Organization classification, including pediatric-type FL and duodenal FL, although these are not discussed. Here, we summarize recent insights into the molecular underpinnings of FL, gain an appreciation of the disease's heterogeneity, and discuss the potential for rational clinical translation.
Tripartite intersection of genetics, epigenetics, and the microenvironment
Genetic landscape and the importance of epigenetic regulation
The compendium of genetic events within the coding region in FL is now well defined (Table 1). The t(14;18) translocation leading to BCL2 overexpression occurs in approximately 85% of patients with advanced-stage disease (compared to a lower incidence in limited-stage FL) and is considered the first genetic event in the oncogenic pathway. A major discovery within the last decade involves the high prevalence of somatic mutations affecting genes with key roles in histone modification and, ultimately, epigenetic regulation.6–8 Mutations in histone methyltransferases (KMT2D, EZH2) and acetyltransferases (CREBBP, EP300) occur in as many as 90% of FL patients. Many patients harbor multiple mutations in these genes, indicating that the consequential downstream epigenetic deregulation is a defining feature of FL,6–8 more so than other NHLs.9
Main classes of mutations in follicular lymphoma
| Mutational class | Frequency | Mutated/altered genes |
|---|---|---|
| Epigenetic modifiers | >90% | KMT2D, CREBBP, EZH2, EP300, HIST1H1B-E |
| Immune regulation | ~28%-40%; 6% | TNFRSF14, CTSS |
| mTOR signaling | >30% | RRAGC, ATP6V1B2, ATP6AP1, SESTRIN1 |
| Cell cycle regulators | ~10%-15% | CDKN2A/B, CCND3, MYC, TP53 |
| Jak/STAT signaling | ~20% | STAT6, SOCS1 |
| NFκB signaling | >15% | CARD11, TNFAIP3, CD79A/B, MYD88 |
| Mutational class | Frequency | Mutated/altered genes |
|---|---|---|
| Epigenetic modifiers | >90% | KMT2D, CREBBP, EZH2, EP300, HIST1H1B-E |
| Immune regulation | ~28%-40%; 6% | TNFRSF14, CTSS |
| mTOR signaling | >30% | RRAGC, ATP6V1B2, ATP6AP1, SESTRIN1 |
| Cell cycle regulators | ~10%-15% | CDKN2A/B, CCND3, MYC, TP53 |
| Jak/STAT signaling | ~20% | STAT6, SOCS1 |
| NFκB signaling | >15% | CARD11, TNFAIP3, CD79A/B, MYD88 |
The mutations in KMT2D, CREBBP, and EP300 are commonly inactivating, whereas EZH2 mutations are gain of function, leading to a disequilibrium at the histone 3 lysine 4 and histone 3 lysine 27 marks and subsequent perturbations in gene expression. These aberrations contribute to lymphomagenesis by exerting broad pleiotropic contributions ranging from locking B cells at the germinal center differentiation stage and switching on B-cell survival signaling pathways to disrupting the tumor- immune cross talk and promoting immune evasion (Figure 1).8,10-13 Aside from mutations that have an impact on histone modifiers, frequent mutations in genes encoding components of chromatin remodeling complexes, such as isoforms of linker histones (HIST1H1B-E; also, H1B-E), also occur in FL.6,14 The functional consequence of these heterozygous mutations adds to the ongoing complex epigenetic reprogramming and gene silencing in FL by impairing chromatin compaction and the three-dimensional genome organization.15

Consequences of the mutations affecting epigenetic regulators and the mTORC1 signaling pathway in FL. (A) Mutations in epigenetic regulators affect respective histone modifications with a convergence on specific downstream functional effects; (B) RRAGC mutations have an impact on the mTORC1 nutrient signaling pathway and are able to enhance B-cell activation even with a reduction in nutrient levels. MHC, major histocompatibility complex.
Consequences of the mutations affecting epigenetic regulators and the mTORC1 signaling pathway in FL. (A) Mutations in epigenetic regulators affect respective histone modifications with a convergence on specific downstream functional effects; (B) RRAGC mutations have an impact on the mTORC1 nutrient signaling pathway and are able to enhance B-cell activation even with a reduction in nutrient levels. MHC, major histocompatibility complex.
Outside of epigenetic dysregulation, mutations or copy number alterations frequently target genes involved in biological pathways ranging from immune recognition (TNFRSF14, CTSS) to signaling pathways such as BCR-NFκB (CARD11, TNFAIP3), Jak/STAT (STAT6), and mTOR (RRAGC, ATP6V1B2, ATP6AP1, SESTRIN1).6,8,14,16–20 There is a convergence of genetic aberrations on the amino-acid-sensing arm of the mTORC1 signaling pathway, emphasizing the significance of aberrant metabolic reprogramming in FL pathogenesis and the possibility that these aberrations allow the lymphoma cells to bypass the normal physiological metabolic checkpoints (Figure 1).19–21 Notably, while the genetic profile between t(14;18)-positive and t(14;18)-negative FL overlaps, there are distinct patterns observed, with a much higher frequency of STAT6 mutations in the latter.22
An important consequence of certain genetic aberrations is their ability to co-opt and reshape the microenvironment into one conducive to lymphoma survival. The impact of mutations or deletions in TNFRSF14, which encodes a bidirectional signaling molecule, is an increase in follicular helper CD4+ T cells (Tfh) infiltration and activation of the tumor stroma.16 Mutations or overexpression of cathepsin S (CTSS) leads to enhanced activation in lymphomas, subsequently increasing antigen-specific CD4+ T-cell activation and infiltration to garner tumor support and promote immune evasion through the exclusion of cytotoxic CD8+ T cells.17,18 CREBBP loss in mouse models and patients' lymphomas show a downregulation of major histocompatibility complex class II, with a more profound downregulation associated with missense mutations within the CREBBP HAT domain compared to truncating protein mutations.8,12,23 EZH2 mutations can affect major histocompatibility complex class I/II downregulation as well as reprogram the cross talk between the lymphoma B cells and the microenvironment by modulating the TME composition.11,24
Despite a better understanding of the genetic landscape, linking the genotype to clinical outcomes, similar to recent advances in DLBCL, is still in its infancy. A recent study identified 3 genotypic subgroups within the umbrella of FL: the first cluster associated with a high burden of aberrant somatic hypermutation, a second with frequent STAT6 and CREBBP mutations, and a third group enriched for KMT2D mutations without the features of the prior clusters.25 Notably, none of these 3 groups coassociated with patient risk or propensity to transformation, although a key limitation was the use of targeted gene sequencing rather than broader genomic approaches to define these genetic clusters.
Tumor microenvironment in FL
The TME in FL is a compelling driver and influencer of lymphoma initiation and progression via dynamic bidirectional cross talk with the lymphoma B cells. Initial studies focused on determining the FL TME composition and spatial organization using flow cytometric and immunohistochemical approaches. The seminal study by Dave et al., nearly 20 years ago, provided early evidence that different TME phenotypes can yield information relevant to a patient's prognosis.26 Continued advances in our understanding of the immune and stromal contexture provide insight into why some patients, compared to others, respond to specific immunotherapies and help identify opportunities to reprogram and redirect the microenvironment toward the lymphoma.
The cellular composition of the FL TME varies spatially and, most likely, also during different stages of the disease, albeit the focus has primarily been on pretreatment specimens. One of the most prominent findings is the increase in specific CD4+ T-cell subsets, such as Tfh cells, regulatory T cells (Treg), and follicular regulatory T cells (Tfr) compared to normal lymph nodes. In FL, the Tfh secrete and trigger certain cytokines (IL-2, IL-4) and chemokines (CXCL12) that favor the growth and survival of FL cells.27,28 Higher proportions of Tregs with an enhanced immune-suppressive capacity that hampers CD8 T-cell activation are prevalent in FL.29 A specialized subset of Tfr cells with features between Tfh and classical Tregs are observed in FL. Not only is there a skew in T-cell composition in the FL microenvironment; there are also variations in functional T-cell states. Persistent antigen stimulation results in the upregulation of multiple inhibitory checkpoint receptors, including PD-1, LAG3, TIM3, and TIGIT, leading to the hijacking of immune-suppressive signaling pathways, CD8+ T-cell dysfunction, and exhaustion induction and tolerance, with these mechanisms promoting an overall tumoral immune evasion.30,31
Higher-resolution technologies such as time-of-flight mass cytometry (CyTOF) and single-cell transcriptomic analyses are providing a more detailed picture of the immune contexture within FL and demonstrate a much greater heterogeneity, plasticity, and complexity in T-cell subsets than previously appreciated. Using CyTOF, the FL niche shows an enrichment for “prematurely aged” T cells that lack the costimulatory molecules CD27 and CD28, and increased numbers of these CD27−/CD28− T cells have been associated with inferior clinical outcomes.32 Recently, using a combination of CyTOF and codetection by indexing (CODEX) for immune characterization and spatial organization, the lack of intrafollicular CD4 expression predicted early treatment failure in FL and could provide a means of identifying high-risk patients.33
The importance of the TME milieu in FL is not limited to T cells. Tumor-associated macrophages in FL contribute to lymphoma growth and are linked to treatment responses and prognosis.34 Mesenchymal stromal cells in the bone marrow become activated and secrete chemokines such as CXCL12 and CXCL13 that promote B-cell homing and have the ability to differentiate into other stromal components, such as fibroblastic reticular cells and follicular dendritic cells, that influence tumor growth.35,36 Indeed, a recent study illustrated that FL B cells can reprogram the cross talk with lymphoid stromal cells, particularly enhancing unique interactions with a subset of CD49a+ fibroblastic reticular cells.37
Genetic heterogeneity and evolution in FL
Longitudinal genetic studies of FL patients from diagnosis to relapse and transformation have taught us the patterns of clonal evolution and the genetic drivers that occur early or later in the disease's development (Figure 2). Every episode of disease (diagnosis, relapse, or transformation) from an individual patient appears to originate from a putative founder population referred to as the common progenitor cell (CPC).6,38,39 This population acts as the lymphoma-propagating reservoir and is genetically characterized by the t(14;18), CREBBP, and KMT2D mutations in most cases. Its persistence and ability to evade therapy and clonally expand over time may explain the frequent relapses and incurable nature of FL. These longitudinal studies have also demonstrated the changing genetic repertoire from indolent to transformed FL, evolving from the CPC with the later acquisition of alterations, including mutations or copy number changes in cell-cycle regulators such as CDKN2A/B, MYC, TP53, immune surveillance (B2M), and NFκB signaling (MYD88, TNFAIP3).6,38,39 Despite an enrichment of these genetic alterations at transformation, they are not easily detected at diagnosis, and therefore predictive biomarkers of incipient FL transformation are still lacking.
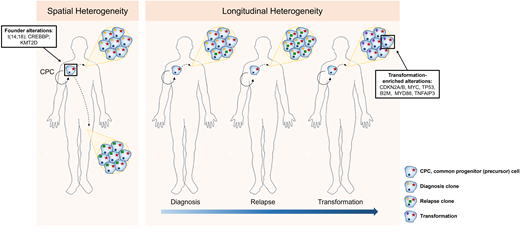
Spatial and longitudinal genetic heterogeneity in FL. Initiating genetic events occur within an ancestral population (CPC), with divergence, acquisition of new genetic events, and subsequent expansion either at different sites of disease involvement or at later time points (relapse or transformation).
Spatial and longitudinal genetic heterogeneity in FL. Initiating genetic events occur within an ancestral population (CPC), with divergence, acquisition of new genetic events, and subsequent expansion either at different sites of disease involvement or at later time points (relapse or transformation).
The stepwise evolution from normal B cells to premalignant states like in situ follicular neoplasia to overt FL and, importantly, where the CPC model fits within this process, remains incompletely defined.40,41 Similarly, the exact temporal order of the acquisition of genetic aberrations continues to be debated, with observations that in some instances the aberrations in epigenetic regulators might occur earlier than the germinal center but unlikely prior to the BCL2 translocation.42,43
Recent studies have highlighted the degree of spatial (different disease-involved sites), genetic, and transcriptional heterogeneity in FL, further highlighting the inability of a single biopsy to capture the biological spectrum within a patient's disease (Figure 2).44,45 Single-cell transcriptomic studies demonstrated that the discordance between lymphomas at spatially distinct compartments correlated with a divergence in CD4 Tfh abundance.45 It is unclear whether the tumor heterogeneity leads to a differential ability to co-opt Tfh cells or whether Tfh cell signals exert selective pressure and act as additional drivers of the lymphoma heterogeneity, presenting a chicken-and-egg situation. Additionally, the higher resolution afforded by single-cell technologies illustrates that, even within a single patient's FL biopsy, malignant B cells exist in a continuum of different B-cell states broader than the traditional view that all the cancerous lymphoma cells in FL are predominantly of germinal center cell origin.46
Window of opportunities for clinical translation
As we gain an improved understanding of the cellular and molecular complexity of FL, are we suitably poised to begin translating this knowledge into new opportunities for patient benefit? Current and future areas of development include the prioritization of targeted therapies to revert the abnormal genomic profiles in addition to identifying and validating robust biomarkers for risk stratification, disease monitoring, and therapy selection.
New targets, new therapeutics
Efforts to therapeutically target and revert epigenetic dysregulation are warranted given the high frequency and driver role in FL pathogenesis. The only US Food and Drug Administration– approved epigenetic therapy for FL is the direct EZH2 inhibitor tazemetostat, the treatment offered in our Clinical Case. Patients with EZH2-mutant lymphomas treated with tazemetostat had superior overall response rates compared to those with EZH2 wild-type lymphomas (overall response rate, 69% vs 35%).47 Potential therapeutics to abrogate the effects of CREBBP- and KMT2D-mutated lymphomas are being explored, including selective CREBBP, HDAC3, and KDM5 inhibitors.23,48,49 Hypothetically, targeting these early driver mutations may lead to eradication of the reservoir CPC population and perhaps reduce the propensity for relapses.
Broadening prognostic and predictive biomarkers
The up-front identification of patients who are at risk of inferior outcomes (prognostic markers) remains an unmet clinical need. Clinical indices like FLIPI, FLIPI-2, and more recently the Protein-RNA Interaction Mapping Assay-Prognostic Index are capable of risk stratifying patients but have not informed treatment decisions.50–52 The first iterations of biology-based prognostic tools, m7-FLIPI (based on integrating the mutation status of 7 genes together with FLIPI score and performance status) and the Protein-RNA Interaction Mapping Assay 23-gene expression-based signature, have been developed.53,54 Both demonstrate feasibility in stratifying patients by risk at diagnosis, although their ability to identify high-risk groups like early progressors remains imperfect and appears highly induction treatment dependent.55 Going forward, improved iterations in biological-based prognostic tools are anticipated.
With an ever-increasing armamentarium of therapeutic options in FL in both the frontline and relapsed settings, there must be a greater emphasis on defining the biological determinants of both treatment response and resistance (predictive markers) to aid in rationalizing who benefits most from specific therapies. At present, EZH2 mutation status remains the only current predictive biomarker at our disposal.
Dynamic disease monitoring
The spatial and temporal heterogeneity of FL has taught us that single biopsies never accurately capture the changing patterns in a patient's disease. The genomic assessment of circulating tumor DNA provides a means of surveying disease heterogeneity and behavior ranging from monitoring therapeutic responses to forecasting treatment failures and detecting disease progression or transformation. Its utility in DLBCL is paving the way for similar areas of exploration in FL, with the role of circulating tumor DNA being evaluated in ongoing FL trials, including PETReA and FOLL19.56–58 The application of this promising approach requires standardization, prospective validation, and correlation with existing modalities like imaging, minimal residual disease monitoring, and other biomarker strategies.
CLINICAL CASE (Continued)
This patient carried the hot-spot mutation at the Y641 residue in EZH2 and attained a partial response with the EZH2 inhibitor tazemetostat. Notably, this epigenetic therapy is approved for relapsed, refractory FL patients with both the EZH2 mutation or the wild type. Although higher response rates are seen in EZH2-mutated patients, the median progression-free survival is similar in both mutated and wild-type groups and, importantly, is short, perhaps indicating that targeting single epigenetic alterations is insufficient for the complete eradication of lymphomas.47 When mutation profiling is performed, this should ideally be undertaken on the biopsy closest to the relapse event, as occasionally the EZH2-carrying clone can be lost during the later stages of a patient's disease course. Presently, there are no other clinical indications for broader genetic sequencing beyond screening for EZH2 mutations in those patients considered for tazemetostat.
Future directions
Despite our current advancements, we are some way from harnessing our growing understanding of FL biology. While there are many, we highlight 2 initial broad priority areas that bridge this chasm and expand our windows of clinical opportunity beyond those already discussed above, including the following:
- 1)
Continued efforts to enhance understanding of FL heterogeneity: There is increased recognition of the importance of different biological layers beyond the genetics of FL. How these vary in specific patient populations and over the course of their disease is still incomplete. For example, it is crucial to understand how the TME varies across clinical phenotypes, how it evolves over time, and how it is reshaped by prior therapies, as this could have major implications in deciding future therapies like bispecific antibodies and chimeric antigen receptor T cells and open up avenues for manipulating the TME to enhance treatment efficacy. This in turn allows the discovery of multidimensional biomarkers coupled with the dynamic tools already discussed earlier.
- 2)
Trial design evolution: With the growing number of therapies, particularly in the relapsed, refractory FL space, therapy selection remains primarily biology agnostic. To improve our ability to get the best available treatment(s) to the right patient group, there is a need to strengthen and routinely incorporate robust parallel correlative studies, ideally developed from the outset alongside our prospective clinical trial designs. This requires cross talk and collaboration between clinical and lymphoma biology investigators and proactive biobanking of appropriate tissue specimens to enable both the discovery and hypothesis-driven evaluation of biomarkers, particularly those linked to therapeutic response or resistance.
Conclusions
Studies in the last decade have shed light on the complex pathogenesis of FL, in particular the tripartite connectivity between the genetics, the epigenetics, and the microenvironment as critical drivers and enablers of this disease. Insights into the (epi)genetic landscape provide opportunities for new ways of therapeutic targeting. Furthermore, these lymphomas are not biologically static, and our future approaches must incorporate ways of tracking and adapting to these changes. Ultimately, this biological heterogeneity forces us to refocus our efforts into defining better prognostic and predictive tools that in turn inform our therapeutic strategies and facilitate further improvements in patient outcomes.
Acknowledgments
Megan Perrett is supported by a Cancer Research UK Accelerator Award Studentship (C355/A28222). Carina Edmondson and Jessica Okosun are funded by Cancer Research UK (C57432/ A22742).
Conflict-of-interest disclosure
Megan Perrett: no competing financial interests to declare.
Carina Edmondson: no competing financial interests to declare.
Jessica Okosun: no competing financial interests to declare.
Off-label drug use
Megan Perrett: nothing to disclose.
Carina Edmondson: nothing to disclose.
Jessica Okosun: nothing to disclose.
