

Abstract
Langerhans cell histiocytosis (LCH) is a rare myeloid neoplasm driven by activating mutations in the MAPK pathway, most commonly BRAF-V600E and MAP2K1. It affects children and adults, with a wide spectrum of clinical presentations ranging from self-limited to multisystem (MS) life-threatening forms. LCH is defined by the accumulation of CD1a+/CD207+ cells in different organs, and patients with liver, spleen, or hematopoietic system involvement have a higher risk of mortality. Patients with neurodegeneration (ND) have devastating outcomes and are resistant to systemic therapies. MS-LCH is treated with risk-adapted therapy, but many patients require multiple salvage regimens that are myelosuppressive and expensive. MAPK inhibitors are increasingly being used, but most patients relapse upon discontinuation of therapy. Here, we review the management of central nervous system disease and how novel cerebrospinal fluid biomarkers might predict patients at high risk of ND who could benefit from early MAPK inhibition. Further, we discuss treatment strategies for refractory/relapsed (R/R) LCH, with a focus on MAPK inhibitors' efficacy and challenges (ie, the unknown): long-term toxicity in children, optimal duration, if they are curative, whether it is safe to combine them with chemotherapy, and their high price tag. Lastly, emerging strategies, such as the new panRAF inhibitor (Day 101) in patients with R/R LCH, ERK1/2 or CSF1R inhibition in patients with MEK1/2 inhibitor resistance, and targeting the microenvironment (checkpoint plus MEK inhibition) or senescent cells (mTOR or BCL-XL inhibitors) in R/R patients, are also examined.
Learning Objectives
Discuss the treatment approach to patients with neurodegeneration and the potential role of novel cerebrospinal fluid biomarkers
Learn how comprehensive genomic profiling at the diagnosis of Langerhans cell histiocytosis can add prognostic information and positively impact clinical care
Describe the promises and caveats of MAP kinase targeted therapies in refractory/relapsed high-risk Langerhans cell histiocytosis
CLINICAL CASE
Federico, a 2-year-old boy, presented with scalp rash (Figure 1A), polyuria, polydipsia, hepatosplenomegaly, pancytopenia, and liver dysfunction. Skin biopsy revealed CD1a+/CD207+ histiocytes (Figure 1B, C), compatible with Langerhans cell histiocytosis (LCH); BRAF-V600E was positive (Figure 1D). Brain magnetic resonance imaging (MRI) showed pituitary stalk thickening, absent posterior bright spot (Figure 1E). Vinblastine/prednisone for 6 weeks were given with no response. Salvage with cladribine/cytarabine followed by oral mercaptopurine (6-MP)/methotrexate led to complete remission (CR) for 8 years.

Clinical, pathological, and radiographic features of LCH with cutaneous toxicities of MAPK inhibitors. (A) Scaly scalp rash in a 2-year-old boy. (B) LCH with a rich inflammatory background including osteoclast-like giant cells, eosinophils, and neutrophils (hematoxylin and eosin). (C) Immunostain showing surface CD1a expression. (D) Mutant-specific BRAF-VE1 immunostain with dark granular cytoplasmic staining in the lesional histiocytes (immunostain 400 × ). (E) Granulomatous CNS-LCH: brain MRI sagittal T1W image lacking posterior pituitary bright spot. (F) Brain MRI in a patient with granulomatous CNS-LCH: axial contrast-enhanced T1W image showing extensive bilateral lesions in the choroid plexus. (G) ND-LCH: axial T2-weighted image showing extensive dentate nucleus and white matter cerebellar neurodegeneration. (H) Skin hyperkeratosis pilaris of the left arm in a patient treated with a BRAF inhibitor. (I) Lobular panniculitis of the right leg in a patient on a MEK inhibitor. (J) Cutaneous squamous cell carcinoma in a adult treated with a BRAF inhibitor.
Clinical, pathological, and radiographic features of LCH with cutaneous toxicities of MAPK inhibitors. (A) Scaly scalp rash in a 2-year-old boy. (B) LCH with a rich inflammatory background including osteoclast-like giant cells, eosinophils, and neutrophils (hematoxylin and eosin). (C) Immunostain showing surface CD1a expression. (D) Mutant-specific BRAF-VE1 immunostain with dark granular cytoplasmic staining in the lesional histiocytes (immunostain 400 × ). (E) Granulomatous CNS-LCH: brain MRI sagittal T1W image lacking posterior pituitary bright spot. (F) Brain MRI in a patient with granulomatous CNS-LCH: axial contrast-enhanced T1W image showing extensive bilateral lesions in the choroid plexus. (G) ND-LCH: axial T2-weighted image showing extensive dentate nucleus and white matter cerebellar neurodegeneration. (H) Skin hyperkeratosis pilaris of the left arm in a patient treated with a BRAF inhibitor. (I) Lobular panniculitis of the right leg in a patient on a MEK inhibitor. (J) Cutaneous squamous cell carcinoma in a adult treated with a BRAF inhibitor.
Introduction
LCH is a rare disorder characterized by expansion of myeloid precursors that differentiate into CD1a+/CD207+ lesions. It affects children and adults with a wide spectrum of clinical manifestations, ranging from single-system (SS) to multisystem (MS) disease. LCH is an inflammatory myeloid neoplasm due to the presence of MAPK-ERK pathway mutations in most cases, beyond BRAF-V600E.1-3 Next-generation sequencing (NGS) showed that BRAF wild-type LCH harbors other mutations, like MAP2K1 (in 15%), KRAS, NRAS, ARAF, MAP3K1, ERBB3,3 kinase fusions (BRAF, NTRK1, ALK), BRAF duplications, insertions, deletions, and PI3K-AKT-mTOR pathway/receptor-colony stimulating factor 1 receptor (CSF1R) mutations3,4 (Figure 2A). LCH incidence ranges from 2.6 to 8.9 cases/million children/year5 and 1-2 cases/ million/year in adults.6
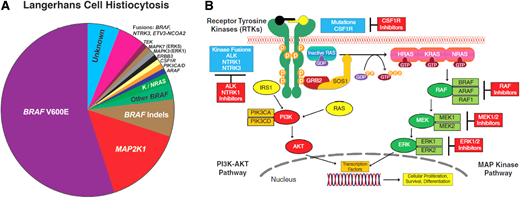
(A) Summary of diverse kinase alterations discovered by next-generation sequencing techniques in LCH in the past 13 years: pie chart illustrating a composite of the diverse kinase alterations driving LCH, many of which are targetable. (B) Diagram of the MAPK and PI3K-AKT signaling pathways: description of the activation of the RAS proteins with annotation of the signaling proteins affected by genetic alterations in the histiocytic neoplasms.
(A) Summary of diverse kinase alterations discovered by next-generation sequencing techniques in LCH in the past 13 years: pie chart illustrating a composite of the diverse kinase alterations driving LCH, many of which are targetable. (B) Diagram of the MAPK and PI3K-AKT signaling pathways: description of the activation of the RAS proteins with annotation of the signaling proteins affected by genetic alterations in the histiocytic neoplasms.
The classification of LCH in children is based on number of involved sites (SS, MS/unifocal or multifocal) and if risk organs (ROs; risk of mortality) like liver, spleen, or the hematopoietic system are involved (Table 1).7 SS/MS disease account for 50% of patients each, and 15% of MS cases are RO+. Bone involvement occurs in 80%; skull is the most common.7 Skin is the second most common, especially in infants, more frequently in the scalp as seborrheic eczema. “Skin-only” LCH has a 60% chance of spontaneous resolution; however, progression to MS disease occurs in 40%.8
Clinical classification of LCH
| Children | |
|---|---|
| Clinical group | Description |
| Multisystem | Two or more systems involved |
| With risk organ involvement | Involvement of liver, spleen, or bone marrow |
| Without risk organ involvement | Without involvement of liver, spleen, or bone marrow |
| Single system | Only 1 system involved |
| Single site | Skin, bone, lymph node, thyroid, thymus |
| Multiple sites | Multifocal bone disease |
| Special site | Skull-base lesion with intracranial extension or vertebral lesion with intraspinal soft tissue extension |
| Pulmonary LCH | Isolated lung disease |
| CNS-LCH | Tumorous lesions |
| Neurodegenerative disease LACI LACS | |
| Adults | |
| Unifocal | Solitary lesion involving any organ |
| Single-system pulmonary | Isolated lung involvement, predominantly smoking related |
| Single-system multifocal | ≥1 lesion involving any organ |
| Multisystem | ≥2 organ/system involvement |
| Children | |
|---|---|
| Clinical group | Description |
| Multisystem | Two or more systems involved |
| With risk organ involvement | Involvement of liver, spleen, or bone marrow |
| Without risk organ involvement | Without involvement of liver, spleen, or bone marrow |
| Single system | Only 1 system involved |
| Single site | Skin, bone, lymph node, thyroid, thymus |
| Multiple sites | Multifocal bone disease |
| Special site | Skull-base lesion with intracranial extension or vertebral lesion with intraspinal soft tissue extension |
| Pulmonary LCH | Isolated lung disease |
| CNS-LCH | Tumorous lesions |
| Neurodegenerative disease LACI LACS | |
| Adults | |
| Unifocal | Solitary lesion involving any organ |
| Single-system pulmonary | Isolated lung involvement, predominantly smoking related |
| Single-system multifocal | ≥1 lesion involving any organ |
| Multisystem | ≥2 organ/system involvement |
Adult LCH usually presents with MS disease. Clinical classification does not differentiate RO separately, due to lack of data on prognostic implications in the targeted therapies era (Table 1). Pulmonary LCH (PLCH) occurs usually in isolation, mostly associated with smoking, and usually responds to smoking cessation.9
Central nervous system LCH
Central nervous system (CNS) LCH can be granulomatous or neurodegenerative. Granulomatous or tumorous lesions account for 10% to 15% of all LCH cases and tend to occur early in the course of the disease. Typical neuroimaging findings include pituitary stalk thickening (Figure 1E), absent posterior pituitary spot, enlargement of the pineal gland, thickening and enhancement of the choroid plexus (1F), or intraparenchymal masses. Depending on the location of the lesions, patients present more frequently with diabetes insipidus (DI), focal seizures, or symptoms of increased intracranial pressure. DI has been reported to occur in up to 24% of patients with LCH and in half of those with MS disease. The diagnosis of DI usually precedes or is concurrent with the diagnosis of LCH in one-third of cases, while in the remaining two-thirds, it is diagnosed as a late sequela.10 Patients with LCH and coexisting DI are prone to developing anterior pituitary dysfunction, with growth hormone deficiency (in up to 50%), precocious or delayed puberty, hypothyroidism, hypogonadism, hypocortisolism, or panhypopituitarism.10
Neurodegenerative LCH (ND-LCH) can present as LCH- associated abnormal CNS imaging (LACI), which includes asymptomatic patients with radiographic findings in up to 24% of all children with LCH, and LCH-associated abnormal CNS symptoms (LACS), which includes patients with abnormal neurocognitive findings.11 Both forms have increased T2-weighted MRI signals in the dentate nuclei of the cerebellum (Figure 1G), basal ganglia, and pons. Long-term neurodegeneration incidence is 1.9% to 11% and is higher in patients with MS disease, DI, previous CNS-risk bone lesions, or BRAF-V600E–mutated LCH. The onset of radiographic or clinical findings can occur with the initial diagnosis of LCH, although it more commonly occurs years (up to 10) after the resolution of LCH. LACS is a neurodegenerative syndrome of variable severity and course. Symptoms in children may initially include ataxia, dysarthria, tremors, behavioral changes, and learning or psychiatric problems. Some patients may develop a progressive cerebellar syndrome, spastic tetraparesis, pseudobulbar palsy, and cognitive deterioration.10,11 Adult patients usually present with cerebellar deficits.9
Whether ND represents active disease or a sequela of prior active disease is not clear. A recent study identified CD1a- negative BRAF-V600E+ myeloid precursors in brain biopsies of patients with ND-LCH. Further, BRAF-V600E+ cells were identified in the blood of patients with ND who had no systemic findings of active LCH12 ; responses to BRAF inhibitors further support this notion.13 Cerebrospinal fluid (CSF) biomarkers could be a promising tool for patients with ND-LCH. BRAF-V600E mutation can be detected in CSF cell-free DNA in only 10% of cases. CSF osteopontin12 and neurofilament light protein (NFL) are elevated in patients with ND14 ; indeed, CSF NFL levels normalized within 9 months in children receiving MAPK inhibitors, with clinical/ radiologic improvement.14 In summary, novel CSF biomarkers might predict patients at high risk of ND, who could potentially benefit from early MAPK inhibition.
Surveillance
A clinical assessment by a neurologist with standardized tests, such as International Cooperative Ataxia Rating Scale (ICARS) and neuropsychological testing, should be performed initially (as soon as radiologic changes suggestive of ND appear on brain MRI) and at regular intervals for better longitudinal assessment and therapeutic decision-making.11 An ICARS score increase of 5 points indicates clinical deterioration, which, together with radiologic progression on MRI, should merit initiation of therapy.11 Patients with symptoms suggestive of hormonal deficits should be assessed and monitored by an endocrinologist for appropriate testing and possible hormonal replacement.11
Treatment of CNS-LCH
There is no standard therapy for CNS-LCH. For children with granulomatous lesions and new-onset DI, systemic treatment with a standard LCH regimen, such as vinblastine/prednisone, is advocated with the goal of preventing ND disease and anterior pituitary dysfunction. Reversal of DI has been achieved only in anecdotal cases, and almost all patients require lifelong replacement therapy with desmopressin. Parenchymal mass lesions of the brain due to LCH may respond to vinblastine/ prednisone, cytarabine, cladribine, or clofarabine.15 For adult granulomatous lesions, cladribine, higher doses of cytarabine, or MAPK inhibitors are preferred.9
Treatment of LACS is challenging and less defined; early treatment in patients with worsening symptoms is recommended. Clinical and radiologic stabilization was previously reported with cytarabine, intravenous immunoglobulin, infliximab, or rituximab.16 A retrospective study of children with LCH on BRAF or BRAF/MEK inhibitor combinations showed favorable clinical and radiologic responses in 12 of 13 children with LACS.13 Patients with radiographic ND lesions without symptoms (LACI) do not require any treatment, as there is no proof that starting treatment would prevent progression to clinical ND (LACS).
In summary, the prevention and management of LACS is quite challenging and requires a multidisciplinary approach. Early start of therapy with MAPK inhibitors, with a close neurologic and neuropsychologic monitoring, is recommended (Figure 3). Novel inhibitors with better CNS penetration are urgently warranted (see section on Day 101). In case of intolerance to, or unavailability of, inhibitor therapy, then treatment with cytarabine or immunoglobulin should be considered.
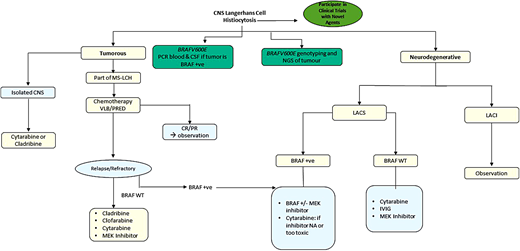
Quality of life and survivorship
Overall morbidity can be significant, resulting in disability in more than half of survivors of MS-LCH. Health-related quality of life, which assesses the patient's perspective of disease burden, has been studied in these patients and was found to correlate closely with morbidity, as measured by professionals. Health- related quality of life parameters were particularly affected by the presence of CNS and lung disease, with an inability to lead independent lives in the most severely affected patients.17
Neurologic problems such as cerebellar ataxia, learning difficulties, and psychological symptoms can develop concurrently or, more often, several years after the diagnosis of LCH. Cerebellar damage may be seen in up to 12% of all patients with LCH, but this increases to 60% in patients with recognized CNS involvement. Neuropsychological sequelae of LCH include intellectual loss, learning deficits, poor school performance, problems with immediate auditory verbal memory, and emotional disturbances.11
Molecular analysis for somatic MAPK-ERK mutations
Immunohistochemical staining/molecular testing for BRAF-V600E should be performed at diagnosis. Quantitative polymerase chain reaction (PCR) or droplet digital PCR is more sensitive than immunohistochemistry or NGS.18 In LCH lesions without BRAF-V600E, NGS for other MAPK mutations is recommended. Peripheral blood (PB) cell-free DNA testing is a good alternative in cases with insufficient lesional tissue, but its correlation with tissue NGS is higher for BRAF-V600E than other mutations.18,19
Clinical implications of BRAF and other MAPK mutations
BRAF-V600E correlated with high-risk (HR) LCH, frontline therapy resistance, DI, ND, and relapse in 315 children.19 However, a clinicogenomic study (377 children) questioned the independent prognostic value of lesional BRAF-V600E, which was associated with HR-MS and skin involvement, CNS-risk bones, and gastrointestinal involvement20 (additive unfavorable prognostic factor in HR-LCH21 ), whereas MAP2K1 mutations associated with SS-bone/skin and less MS disease and BRAF exon 12 deletions correlated with lung involvement20 (in accordance with adult PLCH9 ). Although BRAF-V600E correlated with reduced event-free survival (EFS) in all patients, neither BRAF-V600E nor MAP2K1 mutations were associated with EFS when patients were stratified by disease extent. Furthermore, lesional BRAF-V600E status did not affect outcomes in adult LCH.22
BRAF-V600E allele detection in circulating cell-free (ccf) DNA using digital droplet PCR was investigated in children with BRAF-V600E–mutated LCH. After vinblastine-steroid induction, 7 of 7 nonresponders remained positive for ccf BRAF-V600E compared to 2 of 4 partial responders and 0 of 4 complete responders. Thus, ccf BRAF-V600E is a promising biomarker for monitoring response to therapy for children LCH resistant to frontline chemotherapy.23 More recent data, however, showed that BRAF-V600E+ cells persisted in PB after MAPK inhibitor treatment but were not correlated with clinical response.13 In summary, while BRAF-V600E measurements have been helpful in assessing patient clinical responses, they are not considered independent determinants of LCH outcome.
Frontline therapy of MS-LCH
Children
Risk-adapted treatment led to improved survival for childhood LCH. Current therapy for multifocal and MS-LCH is based on the LCH-III trial, including vinblastine/prednisone for 1 year. Long-term overall survival (OS) for HR (RO+) patients is 85%, while OS in low-risk (LR) disease is >95%.24 Nevertheless, 50% of patients will be refractory or develop reactivations, mostly within 2 years. Despite their excellent survival, LR patients with refractory/relapsed (R/R) LCH have morbidities, including chronic pain, hearing loss, sclerosing cholangitis, pituitary dysfunction, growth retardation, and progressive ND; all of these late sequelae are also very common in HR (RO+) patients. The Histiocyte Society LCH-IV trial (NCT02205762) is trying to optimize frontline therapy outcomes by testing prolonged (12 vs 24 months) and intensifying (± 6-MP) treatment for HR patients and comparing 6- vs 12-month treatment for patients with multifocal bone disease.
Adults
The significant increase in survival of patients with MS-LCH seems to have favored children, with 5-year survival rates of only 70% in adults. Similar treatment guidelines are recommended for adults and children, with some modifications.9 For MS disease, vinblastine-based regimens are effective but cause more neuropathies in adults; thus, cytarabine or cladribine/clofarabine is preferred, although BRAF/MEK inhibitors are increasingly being used. For PLCH, smoking cessation is essential for symptom improvement, followed by observation. Patients with severe disease benefit from cladribine, although BRAF/MEK inhibitors can be considered.9
Treatment of refractory/relapsed LCH
Optimal therapy for patients with R/R LCH is undefined. For LR reactivation, less toxic regimens are effective, including cytarabine/prednisone/vincristine, 6-MP, methotrexate, indomethacin, bisphosphonates, or hydroxyurea7,9 (Figure 4). HR (RO+) R/R patients respond to acute myeloid leukemia–like therapies, including cytarabine, cladribine, and clofarabine. Cladribine (5 mg/m2/d × 5 days) yielded responses in 22% of R/R HR and 62% of LR patients, but only 4% had CR by 6 months.25 However, cladribine is associated with long-term myelosuppression and secondary acute myeloid leukemia.9 Cytarabine (100-170 mg/m2/d) yielded 41% 3-year EFS in children with first relapse or greater.26 High doses of cytarabine (1 g/m2/d) and cladribine (9 mg/m2/d) salvage yielded 5-year OS of 85% in 27 HR-RO+ R/R children but was associated with high treatment-related toxicity.27
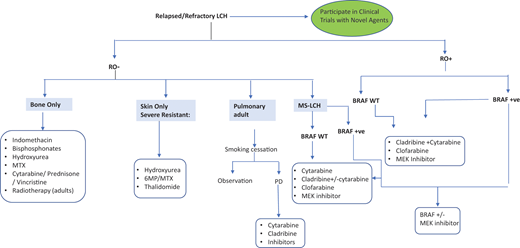
Clofarabine (25 mg/m2/d, 5 days/cycle, for 6 cycles) is also promising, with 1-year PFS of 76% in 11 multiply refractory patients and minimal toxicity.15 Results of a North American Consortium for Histiocytosis prospective study (NCT02425904) testing clofarabine in R/R histiocytoses are pending. Furthermore, patients with multiply refractory HR-RO+ disease were historically treated with hematopoietic stem cell transplant, achieving better outcomes with reduced-intensity conditioning.28 However, since the introduction of targeted therapies, the role of hematopoietic stem cell transplant in LCH has been unclear.9 A summary of the different salvage therapies is shown in Figure 4.
CLINICAL CASE (continued)
Federico's annual brain MRI after 8 years showed new cerebellar white matter lesions (Figure 1G). He was neurologically intact; therefore, he underwent close observation. After 1 year, he developed ataxia/dysarthria with an ICARS score increase of 5 points, and brain MRI showed worsening cerebellar white matter lesions. His symptoms were affecting his quality of life with frequent falls and learning problems at school. Neuropsychologic testing showed a clear cognitive decline. Therefore, he was started on dabrafenib (5.25 mg/kg/d in divided doses) and trametinib (0.025 mg/kg/d) with rapid clinical and radiographic improvement within 2 months.
Rationale for targeted therapies in LCH
Although LCH can be almost universally cured with chemotherapy, major challenges remain. High rates of treatment failure in patients with MS disease, high toxicity of salvage therapies, increased risks of death in HR patients, and increased risk of long-term morbidity for all LCH patients with R/R disease are among those challenges. Thus, more effective and less toxic treatment options are warranted. While chemotherapy drugs are cytotoxic to all rapidly dividing normal and cancerous cells, targeted therapies are cytostatic (block tumor cell proliferation) by acting on specific molecular targets associated with cancer (molecular on/off switch).
Treatment with MAPK inhibitors
Vemurafenib is a BRAF inhibitor that has been approved by the US Food and Drug Administration (FDA) for adults with BRAF-V600E–mutated Erdheim-Chester disease (ECD) and was the first reported targeted therapy to treat refractory MS-LCH in an 8-month old infant.29 Subsequently, an overall response rate (ORR) of 100% was observed in patients with BRAF-V600E– mutated MS and refractory LCH treated with vemurafenib monotherapy in an observational study of 54 children30 and in a small cohort of 4 adults enrolled in a phase 2 basket study (Table 2).31 Dabrafenib, another BRAF inhibitor, given as monotherapy yielded an ORR of 65% in a retrospective study of 20 children with BRAF-V600E–mutated LCH.32 A recently published phase 1/2 study in pediatric patients with R/R BRAF-V600E–mutated LCH showed an ORR of 76.9% (10/13 patients) with dabrafenib monotherapy (NCT01677741), while dabrafenib plus an MEK inhibitor, trametinib (NCT02124772), demonstrated an ORR of 58.3% (7/12) (Table 2).33 Another study of 21 children with MAPK- mutated LCH also reported an ORR of 86% with BRAF or MEK inhibitor monotherapy or combinations.13 MAPK inhibitors have also shown effectiveness in patients with ND-LCH, and patients with early-onset ND achieve better outcomes (Table 2).13,34 Cobimetinib, an MEK1/2 inhibitor, was recently FDA approved for the treatment of adults with histiocytic neoplasms, based on a phase 2 trial in which 20 of 26 (76.9%) patients with different histiocytic neoplasms, including mutations in BRAF, N/KRAS, and MEK1/2, showed a CR or a partial response by fluorodeoxyglucose- positron emission tomography (FDG-PET)35 (Figure 2B).
MAPK-targeted therapies in patients with LCH and other histiocytic disorders
| Disease | Drug name (target) | Dose | N | Age, y | Pathogenic variants | Disease characteristics | Response | Response after DC | Reference |
|---|---|---|---|---|---|---|---|---|---|
| LCH | Vemurafenib* (BRAF) | 8.5-33.8 mg/kg/d | 1 | 0.7 | BRAF-V600E | RR-MS | CR 1/1 | Relapse 1/1 | 29 |
| LCH | Vemurafenib* (BRAF) | 20 mg/kg/d | 54 | 0.2-14 | BRAF-V600E | RR-MS | CR 38/54 PR 16/54 | Relapse 14/30 | 30 |
| LCH | Dabrafenib* (BRAF) Vemurafenib* (BRAF) Trametinib* (BRAF) | NA | 21 | 0.4-21 | BRAF-V600E | RR-MS CNS-ND (13) | CR 4/21 PR 14/21 SD 2/21 PD 1/21 | NA | 13 |
| LCH | Dabrafenib* (BRAF) Trametinib* (BRAF) | D: 5 mg/kg/d T: NA | 4 | 0.1-36 | BRAF-V600E BRAF indel | RR-MS CNS-ND (1) | CR 3/4 PR 1/4 | NA | 34 |
| LCH | Dabrafenib* (BRAF) | 4 mg/kg/d | 20 | 0.6-6 | BRAF-V600E | RR-MS | CR 0/20 PR 14/20 SD 2/20 PD 4/20 | NA | 32 |
| LCH | Dabrafenib* (BRAF) Trametinib* (BRAF) | D: 4.5-5.25 mg/kg/d T: 0.025-0.032 mg/kg/d | 25 D: 13 D+T: 12 | 1-13 | BRAF-V600E | RR-MS (24) CNS-ND (1) | ORR: D: 76.9% D+T: 58.3% | NA | 33 |
| ECD, LCH | Vemurafenib† (BRAF) | 960 mg bid | 26 | 51-74 | BRAF-V600E | Refractory (17/26) CNS (11/26) | CR 2/26 PR 14/26 SD 9/26 | NA | 31 |
| ECD, LCH, RDD, mixed | Cobimetinib‡ (MEK) | 60 mg daily | 18 | 18-80 | BRAF-V600E BRAF MEK1 ARAF MEK2 NRAS WT BRAF | Refractory and/or multisystem and/or brain and/or cardiac involvement | CR 13/18 PR 3/18 SD 1/18 ND 1/18 | NA | 35 |
| Disease | Drug name (target) | Dose | N | Age, y | Pathogenic variants | Disease characteristics | Response | Response after DC | Reference |
|---|---|---|---|---|---|---|---|---|---|
| LCH | Vemurafenib* (BRAF) | 8.5-33.8 mg/kg/d | 1 | 0.7 | BRAF-V600E | RR-MS | CR 1/1 | Relapse 1/1 | 29 |
| LCH | Vemurafenib* (BRAF) | 20 mg/kg/d | 54 | 0.2-14 | BRAF-V600E | RR-MS | CR 38/54 PR 16/54 | Relapse 14/30 | 30 |
| LCH | Dabrafenib* (BRAF) Vemurafenib* (BRAF) Trametinib* (BRAF) | NA | 21 | 0.4-21 | BRAF-V600E | RR-MS CNS-ND (13) | CR 4/21 PR 14/21 SD 2/21 PD 1/21 | NA | 13 |
| LCH | Dabrafenib* (BRAF) Trametinib* (BRAF) | D: 5 mg/kg/d T: NA | 4 | 0.1-36 | BRAF-V600E BRAF indel | RR-MS CNS-ND (1) | CR 3/4 PR 1/4 | NA | 34 |
| LCH | Dabrafenib* (BRAF) | 4 mg/kg/d | 20 | 0.6-6 | BRAF-V600E | RR-MS | CR 0/20 PR 14/20 SD 2/20 PD 4/20 | NA | 32 |
| LCH | Dabrafenib* (BRAF) Trametinib* (BRAF) | D: 4.5-5.25 mg/kg/d T: 0.025-0.032 mg/kg/d | 25 D: 13 D+T: 12 | 1-13 | BRAF-V600E | RR-MS (24) CNS-ND (1) | ORR: D: 76.9% D+T: 58.3% | NA | 33 |
| ECD, LCH | Vemurafenib† (BRAF) | 960 mg bid | 26 | 51-74 | BRAF-V600E | Refractory (17/26) CNS (11/26) | CR 2/26 PR 14/26 SD 9/26 | NA | 31 |
| ECD, LCH, RDD, mixed | Cobimetinib‡ (MEK) | 60 mg daily | 18 | 18-80 | BRAF-V600E BRAF MEK1 ARAF MEK2 NRAS WT BRAF | Refractory and/or multisystem and/or brain and/or cardiac involvement | CR 13/18 PR 3/18 SD 1/18 ND 1/18 | NA | 35 |
D, dabrafenib; NA, not available; ND, neurodegeneration; PD, progressive disease; PR, partial response; RDD, Rosai-Dorfman disease; SD, stable disease; T, trametinib; WT, wild-type.
Drug is not FDA approved for this indication.
Drug is FDA approved for ECD with BRAF-V600E mutation.
Drug is FDA approved for adults with histiocytic neoplasms.
Data on frontline inhibitor therapy in LCH are emerging in adults and children.36,37 Eighteen patients (ages 0.2-45 years) with histiocytoses, including LCH, received frontline MAPK inhibitors. All had favorable responses with a median treatment duration of 2 years. Inhibitors were well tolerated; 5 patients with SS-LCH discontinued therapy and remain in CR off therapy.37
Whether combination therapy with BRAF/MEK inhibitors has greater benefits in patients with BRAF-V600E–mutated LCH compared with BRAF inhibitor monotherapy remains unknown.33 However, combination therapy seems to reduce resistance/ toxicity and improves outcomes in adults and pediatric patients with different types of BRAF-V600E mutant disease.33
CLINICAL CASE (continued)
At 2 years after starting inhibitor therapy, Federico comes back with a 4-week history of bilateral leg pain, myalgias, and frequent falls. Recurrent LCH was ruled out with positron emission tomography/computed tomography. Serum creatine kinase (CK) was high, compatible with mild trametinib-induced rhabdomyolysis. Therefore, trametinib was held and dabrafenib monotherapy continued. After 1 month, his muscular symptoms disappeared and serum CK normalized, and thus trametinib was restarted at two-thirds of his previous dose. Since then, he has been tolerating well his double inhibitors without major side effects.
Caveats of MAPK-targeted therapies
MAPK inhibition in patients with histiocytoses is usually well tolerated. The most common toxicities of BRAF inhibitors include dermatologic (hyperkeratosis pilaris [Figure 1H], migratory panniculitis [Figure 1I], and photosensitivity), fever, vomiting, cough, renal/liver dysfunction, fatigue, constipation, prolonged QT, and joint pain33 ; uveitis was reported in adults. Most adverse events are grade 1 or 2. Toxicities from MEK inhibitors include fever, fatigue, nausea, diarrhea, decreased neutrophil/ platelet counts, skin rash/infections, and rhabdomyolysis; in adults, these include retinopathy, retinal vein occlusion, and decreased ejection fraction.38 Thyroid cancer and leukemia have been reported in adult LCH treated with vemurafenib.31 Further, cutaneous spinocellular carcinoma/squamous cell carcinoma (Figure 1J)/basal cell carcinoma and myeloproliferative syndromes were seen in adult patients with ECD after MAPK inhibitors.38 There are no pediatric reports of second malignancies with MAPK inhibitors, and their long-term toxicities in this population are unknown.
Day 101 (tovorafenib) is a type II panRAF inhibitor that does not cause the severe dermatologic, cardiac, or ophthalmologic toxicities of other RAF/MEK inhibitors. It results in potent inhibition of BRAF-V600E mutations and, in contrast to type I RAF inhibitors (vemurafenib, dabrafenib), inhibits both wild-type BRAF and CRAF/RAFI kinase, as well as hyperactivated signaling resulting from BRAF fusions, including KIAA1549:BRAF fusion.39 In vitro studies demonstrated that Day 101 inhibits phosphorylated ERK signaling and cell proliferation in cell lines with high MAPK activity. Interestingly, Day 101 does not cause paradoxical activation of wild-type RAF kinase dimers in cells with moderate RAS activity; thus, there is less risk of skin rash/cancer.39 An upcoming Children's Oncology Group trial (NCT05287295) will test the efficacy/safety of Day 101 in children, adolescents, and young adults with R/R LCH.
Higher grades (3 or 4) are reversible with dose reduction or inhibitor discontinuation. Successful rechallenge of inhibitor combination was reported in an adult patient with melanoma after trametinib-induced rhabdomyolysis.40 It is common practice in adults, in case of toxicity, to reduce the dose or have intermittent dosing/treatment holidays.9 Patients on BRAF inhibitors need dermatology follow-up to monitor for skin rash/cancer, while patients on MEK inhibitors need, in addition to dermatology, echocardiogram/electrocardiogram and ophthalmology follow-up.
Not all mutations respond to inhibition; thus, evaluating the mutational landscape of patients with LCH before inhibitor therapy is essential. BRAFN486_P490indel mutation is resistant to dabrafenib/vemurafenib but responds to trametinib34 or sorafenib.41 Further, RAF-dependent MAP2K1 mutations respond to RAF inhibitors,42 but RAF-independent mutations (MAP2K1p.L98_K104>Q) are resistant to trametinib.43 Ulixertinib (oral ERK1/2 inhibitor) recently led to CR/partial response in 3 of 4 adults with histiocytic neoplasms (including LCH) with class 3 MAP2K1 mutation (exon 3 p.E102-1103 in-frame deletion) who progressed after MEK inhibition44 (see Figure 2B).
MAPK inhibitors modulate the differentiation and function of LCH cells rather than eradicate precursors, like chemotherapy does; thus, they are not curative. The BRAF-V600E+ cells persist in PB after MAPK inhibition, although this was not correlated with clinical responses.13,34,45,46 Eight of 9 infants receiving salvage vemurafenib/chemotherapy responded without toxicity. Nevertheless, 5 of 8 patients relapsed after discontinuing vemurafenib and required vemurafenib maintenance.46
BRAF/MEK inhibitors are substrates of P-glycoproteins, and their efflux by the blood-brain barrier leads to limited drug levels within the CNS.47 Patients with unfavorable CNS responses to BRAF inhibitor monotherapy in BRAF-V600E–mutated ECD respond to combined BRAF/MEK inhibition.48 In contrast to all approved MEK/BRAF inhibitors, Day 101 has greater CNS penetration.39
Optimal duration of MAPK inhibitors is unknown, and most patients (75%) will relapse upon treatment discontinuation,30,38 but inhibitor reintroduction is usually effective. Lastly, although effective, MAPK inhibitors carry a high price tag. Data are lacking on their cost-effectiveness in patients with histiocytoses (Table 3).
Price summary of MAPK inhibitors with regimens/doses
| Drug | Regimen-dose/cost: USD/month | |
|---|---|---|
| Adults | Children (weight = 20 kg) | |
| Vemurafenib | 480-960 mg bid/$6500-$12 500 | 20 mg/kg/d div bid/$2600 (200 mg bid) |
| Dabrafenib | 75-150 mg bid/$8500-$16 500 | 5.25 mg/kg/d div bid/$5500 (50 mg bid) |
| Trametinib | 1-2 mg qd/$8400-$16 800 | 0.025 mg/k/d qd/$4200 (0.5 mg/d) |
| Cobimetinib | 20-60 mg qd × 21 of 28-day cycle/$10 000-$30 000 | 1 mg/kg/d $10 000-$13 000 (20-25 mg/d) |
| Drug | Regimen-dose/cost: USD/month | |
|---|---|---|
| Adults | Children (weight = 20 kg) | |
| Vemurafenib | 480-960 mg bid/$6500-$12 500 | 20 mg/kg/d div bid/$2600 (200 mg bid) |
| Dabrafenib | 75-150 mg bid/$8500-$16 500 | 5.25 mg/kg/d div bid/$5500 (50 mg bid) |
| Trametinib | 1-2 mg qd/$8400-$16 800 | 0.025 mg/k/d qd/$4200 (0.5 mg/d) |
| Cobimetinib | 20-60 mg qd × 21 of 28-day cycle/$10 000-$30 000 | 1 mg/kg/d $10 000-$13 000 (20-25 mg/d) |
In summary, MAPK inhibitors are quite effective in high-risk LCH patients (ie, those with RO+ MS disease who are refractory or multiply relapsed and had failed more than 1 salvage chemotherapy regimen). Low-risk patients (multifocal bone or MS-RO–) with R/R disease respond to chemotherapy or other agents (Figure 4) and do not require inhibitor therapy. This is due to the unknown optimal duration of these drugs and the risk of indefinite and unnecessary treatment for mild disease. In addition, these inhibitors are not indicated in SS-LCH that can resolve spontaneously, or in LR-LCH (MS-RO–) or MS-LCH-RO+ and rapid early responders, in whom OS rates are at 85% with conventional chemotherapy.24
Emerging therapies
Beyond MAPK pathway inhibitors
Alpelisib, a PI3K inhibitor, led to complete metabolic (PET) and clinical response in an adult patients with LCH who was harboring the M1043V-PI3K mutation.49 A patient with refractory ECD carrying CSF1RR549-E554delinsQ had a sustained CR after treatment with pexidartinib, a CSF1R inhibitor50 ; clinical trials in LCH are warranted.
Combination therapy with PD-1/PD-L1 inhibitors
The inflammatory background of the LCH lesion could be another treatment target. Treating BRAF-V600E+ mice with anti–PD-1 antibodies significantly reduced the disease burden, while MEK inhibitors combined with anti–PD-1 treatment were synergistic, causing a decrease in infiltrating myeloid/lymphoid cells and a restored function of CD8+ T cells51 ; human trials are clearly needed.
Targeting senescent cells
In vitro inhibition of the mTOR pathway with rapamycin (sirolimus) reduced inflammatory cytokines and the differential potential toward mononuclear phagocytes in bone marrow BRAF-V600E+ CD34+cells. It also improved organomegaly and inflammatory infiltration of involved organs, but the apoptosis of BRAF-V600E+ cells did not increase.52 Sirolimus (+ prednisone) induced objective responses in adults with refractory ECD53 and was effective as monotherapy in children with refractory Rosai-Dorfman disease (another non-LCH disorder).54 Prospective trials of sirolimus in patients with LCH is worth consideration, particularly as an exit strategy after inhibitor discontinuation. Lastly, direct targeting at senescence by treating the LCH cells with ABT-263, a BCL-XL inhibitor, could increase their apoptosis and be another promising strtategy.55
Summary
Targeted therapy with MAPK inhibitors is particularly helpful for high-risk LCH that is refractory to treatment or progressive (and possibly fatal). With the current state of knowledge, this remains as the main indication for these drugs. The use of inhibitors in ND-LCH is promising but needs more validation in rigorous clinical trials. The risks associated with these inhibitors are the inability to discontinue treatment, high cost, and almost fully unknown potential for long-term side effects, especially in children. Prospective trials testing novel inhibitors are under way. International collaboration is required to harmonize LCH response criteria during the inhibitors era. Funding agencies and drug companies should support LCH research efforts to improve outcomes for patients, particularly in countries with limited resources.
Acknowledgments
The author is grateful to all patients and their families, as well as to his family for their support in the writing of this manuscript. The author also thanks Drs Marta Wilejto, Benjamin Durham, Eli Diamond, and Jennifer Picarsic for allowing use of their outstanding figures. In addition, the author is very grateful to Dr Sheila Weitzman for her wonderful mentorship and to Connie Grillo for the excellent editorial assistance. A warm thanks also to Denise Stregger and Bina Gandhi for their great coordination of the histiocytosis clinic.
Conflict-of-interest disclosure
Oussama Abla: no competing financial interests to declare.
Off-label drug use
Oussama Abla: Dabrafenib, vemurafenib and trametinib are not FDA approved for LCH.
