

Abstract
Chronic lymphocytic leukemia (CLL) patients who are refractory to both Bruton's tyrosine kinase and B-cell/CLL lymphoma 2 (BCL2) inhibitors face a significant treatment challenge, with limited and short-lasting disease control options. This underscores the urgent need for novel therapeutic strategies. Immunotherapy has emerged as a promising approach to address this unmet need, offering the potential for durable remissions and improved patient outcomes. Historically, allogeneic stem cell transplantation has been used for high-risk CLL patients, demonstrating promising survival rates. However, its applicability is limited by high treatment-related mortality and chronic graft-versus-host disease, especially in older and frail patients. Chimeric antigen receptor (CAR) T-cell therapy is gaining attention for its potential in relapsed/refractory CLL. Early clinical trials have shown that CAR T cells can induce durable remissions, with encouraging overall response rates in heavily pretreated patients. Additionally, bispecific antibodies are being explored as immunotherapeutic strategies, showing promising preclinical and early clinical results in targeting CLL cells effectively. One of the major challenges in CLL treatment with T-cell–based therapies is the acquired T-cell dysfunction observed in patients. To overcome these limitations, strategies such as combining targeted agents with cellular immunotherapies, modifying CAR designs, and incorporating immunomodulatory compounds into the manufacturing process are being investigated. These innovative approaches aim to enhance T-cell engagement and improve outcomes for CLL patients, offering hope for more effective and sustainable treatments in the future.
Learning Objectives
Understand therapeutic potential and toxicities of existing and emerging T-cell–based therapies in CLL
Understand the challenges of T-cell–based therapies in CLL and potential strategies to overcome these
CLINICAL CASE
A 51-year-old man was diagnosed with chronic lymphocytic leukemia (CLL) in late 2014, presenting with a white blood cell count of 13 000/µL, a platelet count of 40 000/µL, a hemoglobin level of 11 g/dL (110 g/L), hepatosplenomegaly, diffuse lymphadenopathy, and a genetic profile showing 11q deletion, 13q deletion, and unmutated immunoglobulin heavy chain variable region. He received fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy from February to July 2015, which initially improved his thrombocytopenia but was followed by gradual worsening. In September 2016 he enrolled in an acalabrutinib clinical trial, responding well for several years before progressing and discontinuing the study in April 2020. Next-generation sequencing revealed a Bruton's tyrosine kinase (BTK) mutation (C481S). He then started commercial venetoclax with rituximab on 22 April 2020, continuing 400 mg/d for 2 years. By 24 June 2024, he was progressing again, with worsening lymphocytosis (white blood cell count increased from 4840/µL on 18 March 2024 to 29 220/µL on 24 June 2024), worsening thrombocytopenia, lymphadenopathy, and splenomegaly. To gain rapid control, he started pirtobrutinib in conjunction with venetoclax. The medical team is currently evaluating options for venetoclax taper and lisocabtagene-maraleucel while also testing for evolution of mutations in the CLL cells. This case highlights the challenges of managing CLL with evolving genetic abnormalities and demonstrates both the potential benefits and limitations of targeted therapies in treating this complex disease.
Introduction
Patients with CLL who are refractory to both BTK and BCL2 inhibitors represent a significant unmet clinical need. A wide search for alternative targeted therapies is ongoing, including noncovalent BTK inhibitors (BTKis) like pirtobrutinib and BTK degraders for BTKi-refractory cases. However, for these agents resistance due to novel mutations remains a challenge.1,2 For BCL2 inhibitor alternatives, other BCL2 family inhibitors targeting myeloid cell leukemia-1 or BCL-extra large have been studied, but specific toxicities have hampered progress.3,4 The efficacy of T-cell–based therapies in relapsed/refractory CLL has been demonstrated through allogeneic stem cell transplantation (alloSCT). To mitigate transplant-related toxicities, autologous T-cell approaches, such as chimeric antigen receptor (CAR) T cells and bispecific antibodies (bsAbs), are under investigation, with CAR T cells approved for use in the United States. However, T-cell dysfunction in CLL poses a major challenge to these strategies. This review examines the available data on treatment modalities aimed at directing T cells to CLL cells, their limitations, and future directions to enhance T-cell engagement in CLL treatment (Figure 1).
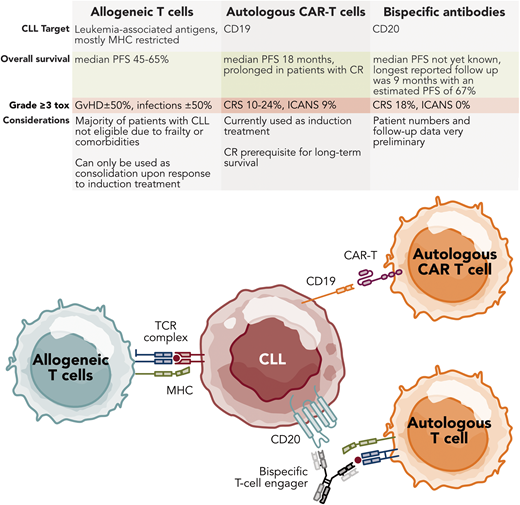
Autologous T-cell-based therapies for CLL. Professional illustration by Somersault18.24.
Autologous T-cell-based therapies for CLL. Professional illustration by Somersault18.24.
Allogeneic SCT
CLL typically has an indolent, chronic course over a patient's lifetime, but it can behave more aggressively if high-risk features of the disease exist, like TP53 mutation/del17p. Historically, chemoimmunotherapy (CIT) did not produce durable responses in patients with high-risk features, and alloSCT was utilized as a potentially curative treatment if an appropriate donor was found because the graft-versus-leukemia effect could outweigh the risks of graft-versus-host disease (GVHD) and life-threatening infections. Limited prospective trials have shown that alloSCT can achieve a durable remission in a significant proportion of patients, including those with high-risk, CIT refractory, and del(17p)/TP53-mutated disease, with reported 5-year overall survival (OS) of about 60% to 65%.5,6 However, nonrelapse/treatment-related mortality can be up to 25% even with reduced-intensity conditioning regimens, and about 50% chronic extensive GVHD incidence can be seen.7-9 At the 10-year median follow-up, 1 prospective trial has reported, nonrelapse mortality of 20% and OS of 52.10 In addition, data from the European Society for Blood and Marrow Transplantation registry showed that even after 10 years, life expectancy remained lower than age-matched controls due to transplant-related morbidity.11 The risk: the benefit ratio of alloHCT has slowly been shifting over the years, even for high-risk CLL patients, due to the availability of improved treatment options, particularly BTKis and BCL2is, either given as a continuous single-agent or a time-limited treatment in combination with each other or with CD20 monoclonal antibodies. However, if patients become refractory or relapse after BTKi and BCL2i and are considered fit, achieve remission, and have a suitable donor identified, then a nonmyeloablative/reduced intensity alloHSCT approach should be considered. Novel schemes, including “postallo” cyclophosphamide, and novel immunosuppressive agents have led to a wider availability of donors, including matched unrelated and haplo-identical donors, with acceptable toxicity.12 With CAR T cells now available in the United States and more access to autologous cellular therapy trials, the decision for alloSCT is complex. However, it is important to note that in most parts of the world, CAR-T therapy is not yet available for CLL, and clinical trials are not easily accessible, particularly in countries facing economic challenges. This disparity in access to advanced therapies further complicates treatment decisions globally.
Achieving a deep remission with manageable toxicity is crucial for the success of alloSCT. Therefore, the risks of alloSCT must be weighed against the patient's health and comorbidities. If the likelihood of remission is high and the risks are acceptable, consolidation with alloSCT should be considered, particularly in younger, fit patients with high-risk disease. In regions where CAR-T therapy is not an option, alloSCT may remain the primary curative approach for eligible patients with high-risk CLL.
The decision-making process should consider not only the patient's clinical status and disease risk but also the availability of advanced therapies like CAR T cells in the patient's location. This global perspective is essential when considering treatment options and potential outcomes for CLL patients worldwide.
In summary, alloSCT can provide long-term remissions in patients with high-risk CLL, particularly when disease control is achieved prior to transplant. Patient selection remains challenging due to immune-related toxicities such as infections and GVHD, making older adult and frail patients, who compose the majority of CLL cases, poor candidates for this procedure. Alternative treatments are urgently needed, especially for patients refractory to both BTKis and BCL2is.
CAR T-cell therapy
The graft-versus-leukemia effects underlying the therapeutic activity of alloSCT imply that CLL can be effectively targeted in a cellular immune response. This has inspired the development of strategies to harness autologous T cells against leukemia cells (Table 1). The proof of concept that CD19 CAR T cells can produce durable remissions, and even be potentially curative, in multiple relapsed CLL came from 2 University of Pennsylvania patients.13,14 The persistence of CTL019 CAR T cells in complete-responding patients at the last follow-up more than 10 years after infusion was reported.15 These patients were part of an early-phase study that included 14 CLL patients.14 The overall response rate (ORR) was 57%, including 4 complete remissions (CRs) and 4 partial remissions (PRs) without an obvious relationship between dose and response or toxicity. Achieving a CR was highly correlated with long-term progression-free survival (PFS). A follow-up study of 42 patients showed an ORR of 44% with a CR of 28%.16 Median PFS in the CR patients was 40.2 months, otherwise only 1 month. Another phase 2 study at Memorial Sloan Kettering Cancer Center evaluated a second-generation anti-CD19 CAR T-cell construct in 8 relapsed CLL patients, with an ORR of 12% and no CRs.17 One PR lasting 6 months was reported. In a phase 1/2 clinical trial conducted by the Fred Hutchinson Cancer Research Center, 49 patients with relapsed/refractory CLL and/or richter transformation (RT) after failing ibrutinib therapy were treated with the CD19 CAR T-cell therapy JCAR014, either with or without concurrent ibrutinib.18,19 With a median follow-up of 6.5 years, the ORR and CR were 70% and 17%, respectively, and the median PFS was 8.9 months. Factors associated with improved outcomes included achieving CR by positron emission tomography and computed tomography and undetectable MRD by multiparameter flow cytometry and next-generation sequencing on day plus 28 post infusion. A higher peak expansion of CD8+ and CD4+ CAR T cells and longer CAR T-cell persistence were also correlated with longer PFS. The longest measured persistence of CAR T cells was 86 months.20 The TRANSCEND CLL 004 trial evaluated lisocabtagene-maraleucel (liso-cel) in 117 patients with relapsed/refractory CLL who had received at least 2 prior lines of therapy, including a BTKi.21 In the primary efficacy analysis set of 49 patients who had progressed after a BTKi, failed a BCL2i, and been treated at the recommended phase 2 dose, the rate of CR was statistically significant at 18% (n = 9; 95% CI, 9-32; P = .0006). The ORR was 43% while the undetectable MRD rate (at a sensitivity of <10−4) was 63% in blood and 59% in marrow. The median duration of response was 35.3 months in this subset (not reached for CR patients), and the median PFS was 11.9 months (not reached for CR patients and 26.2 months for PR patients) with a median OS of 30.3 months (not reached for CR and PR patients).
CAR T-cell therapy trials in relapsed/refractory CLL
| Study | Product | Sample size (enrolled/treated) | Median age (years) | Median prior lines of therapy | ORR (%) | CRR (%) | Median PFS (months) | mOS (months) | Grade 3-5 CRS (%) | Grade 3-5 neurotoxicity (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Porter et al14 | CTL019 (tisagenlecleucel) | 23/14 | 66 | 5 | 57 | 28 | 7 | 29 | 43 | 7 |
| Frey et al16 | CTL019 (tisagenlecleucel) | 42/38 | 62 | 3.5 | 44 | 28 | 1.8 | 64 | 24 | 8 |
| Brentjens et al17 | 2nd-generation CD19 CAR T | 8/8 | 68 | 2-3 | 12 | 0 | Not reported | Not reported | 50 | Not reported |
| Siddiqi et al21 | JCAR017 (lisocabtagene-maraleucel) | 137/117 | 65 | 5 | 47 | 18 | 17.9 | 43.2 | 9 | 19 |
| Liang et al20 | JCAR014 | 49/49 (7 with prior Richter's transformation, 2 with current RT; 30 without ibrutinib and 19 with concurrent ibrutinib) | 61 | 5 | 70 | 17 | 8.9 | 25 | 14 | 27 |
| Study | Product | Sample size (enrolled/treated) | Median age (years) | Median prior lines of therapy | ORR (%) | CRR (%) | Median PFS (months) | mOS (months) | Grade 3-5 CRS (%) | Grade 3-5 neurotoxicity (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Porter et al14 | CTL019 (tisagenlecleucel) | 23/14 | 66 | 5 | 57 | 28 | 7 | 29 | 43 | 7 |
| Frey et al16 | CTL019 (tisagenlecleucel) | 42/38 | 62 | 3.5 | 44 | 28 | 1.8 | 64 | 24 | 8 |
| Brentjens et al17 | 2nd-generation CD19 CAR T | 8/8 | 68 | 2-3 | 12 | 0 | Not reported | Not reported | 50 | Not reported |
| Siddiqi et al21 | JCAR017 (lisocabtagene-maraleucel) | 137/117 | 65 | 5 | 47 | 18 | 17.9 | 43.2 | 9 | 19 |
| Liang et al20 | JCAR014 | 49/49 (7 with prior Richter's transformation, 2 with current RT; 30 without ibrutinib and 19 with concurrent ibrutinib) | 61 | 5 | 70 | 17 | 8.9 | 25 | 14 | 27 |
An important question is whether pre–CAR-T parameters can be identified to predict therapeutic efficacy. In a preliminary exploratory analysis from TRANSCEND CLL 004, Wierda and colleagues recently showed that a lower disease burden correlated with a higher chance of achieving a response and that lisocel was effective regardless of high-risk features of CLL.22 Based on data from the original CTL019 trial, Fraietta et al revealed that clinical efficacy was primarily associated with T-cell characteristics rather than disease factors.23 Premanufacturing T cells from nonresponders showed upregulation of exhaustion, activation, glycolysis, T-memory stem cells and apoptosis genes, while responders' leukapheresis products contained more CD8+ cells. The infused CAR T cells' functionality, determined by in vivo expansion and persistence, was crucial for therapeutic efficacy. Responding patients' CAR T cells exhibited early memory differentiation profiles and decreased aerobic glycolysis dependence. The expression of inhibitory immune checkpoints on infused CAR T cells also impacted outcomes, with nonresponders showing higher levels of CTLA-4, TIM-3, and LAG-3. Additionally, a higher mitochondrial mass in CAR T cells were found to correlate with complete responses.24
Common toxicities of CAR T-cell therapy for CLL include cytokine release syndrome (CRS) and neurological events. CRS occurs in 63% to 85% of patients, with 9% to 24% experiencing severe (grade ≥3) events. Immune effector cell–associated neurotoxicity syndrome (ICANS) affects around 20% to 21% of CLL patients, with severe cases (grade 3 or higher) occurring in approximately 9% of patients.25
In summary, a one-time infusion of CAR T cells shows promise in relapsed/refractory CLL. A CR and long-term CAR T-cell persistence are crucial for durable remissions, thus far achieved in a minority of patients. While efficacy is encouraging in responders, CRS and ICANS remain significant challenges early post cells. The US Food and Drug Administration recently granted accelerated approval to liso-cel for CLL patients who have relapsed after 2 or more lines of therapy, including BTKis and BCL2is.
BsAbs and bispecific T-cell engager immunotherapy
BsAbs and bispecific T-cell engagers (BiTEs) are emerging as promising therapeutic strategies for B-cell malignancies. These agents function by simultaneously binding to tumor-associated antigens and T cells, thereby facilitating T cell–mediated cytotoxicity against cancer cells. Blinatumomab, a BiTE targeting CD19 on B cells and CD3 on T cells, has demonstrated significant preclinical efficacy in CLL. Blinatumomab has exhibited cytotoxicity against CLL cells in vitro, effectively inducing autologous T-cell killing of CLL cells with similar efficacy in both treatment-naive and relapsed/refractory CLL cell lines.26 Clinically, blinatumomab has shown efficacy in a case of refractory RS as a bridge to alloSCT.27 This study demonstrated debulking of coexisting CLL with undetectable minimal residual disease (UMRD) in some patients among the Richter's syndrome cohort treated with blinatumomab. While comprehensive clinical data on blinatumomab in CLL are still limited, these observations strongly suggest that CLL cells are indeed sensitive to this therapy.
Epcoritamab (GEN3013), a bsAb targeting CD3 on T cells and CD20 on B cells, has also shown promising preclinical and early clinical results in CLL. Epcoritamab forms a trimeric complex with CD20 and CD3, leading to T-cell activation, expansion, and subsequent killing of CD20+ B cells. In vitro studies have demonstrated epcoritamab's significant cytotoxic activity against primary CLL cells from both treatment-naive and BTKi-treated patients, with cytotoxic activity that is independent of CD20 expression levels on CLL cells. Notably, the combination of epcoritamab with venetoclax induced superior killing of CLL cells compared to either agent alone and was particularly effective in samples from patients whose condition progressed while receiving BTKi therapy.28 In an ongoing phase 2 clinical trial, epcoritamab demonstrated an ORR of 62% in patients with relapsed/refractory CLL, with 33% obtaining a CR. Interestingly, response rates were similar between patients with high-risk disease, including TP53 aberrations, exposed double-targeted agents, and immunoglobulin heavy chain variable region unmutated disease. The primary toxicity observed was low-grade recurrent CRS, with a small minority accompanied by low-grade ICANS.29 The protocol has been amended to lower CRS risks and include an additional step-up dosing.30Table 2 presents a comprehensive list of current multispecific antibodies undergoing clinical evaluation for CLL and/or small lymphocytic lymphoma (SLL).
Multispecific T-cell engagers in clinical trials for CLL and/or SLL
| NCT number | Multispecific T-cell engager | Target | Title | Status | Intervention |
|---|---|---|---|---|---|
| NCT04623541 | Epcoritamab | CD20 | Safety and Efficacy Study of Epcoritamab in Subjects With Relapsed/Refractory Chronic Lymphocytic Leukemia and Richter's Syndrome | Recruiting | Monotherapy or combination therapy: epcoritamab + venetoclax epcoritamab + lenalidomide epcoritamab + R-CHOP (ie, rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine (Oncovin), and prednisone) |
| NCT06564038 | AZD0486 | CD19 | A Study of AZD0486 Monotherapy or in Combination With Other Anti-Cancer Agents for Mature B-Cell Malignancies | Not yet recruiting | AZD0486 + acalabrutinib, prednisone, rituximab |
| NCT04763083 | NVG-111 | ROR1 | First in Human Study of NVG-111 in Relapsed/Refractory ROR1+ Malignancies Conditions | Recruiting | Monotherapy |
| NCT00889408 | DT2219ARL | CD19 and CD22 | DT2219ARL for Relapsed or Refractory CD19 (+), CD 22 (+) B-Lineage Leukemia or Lymphoma | Completed | Monotherapy |
| NCT02568553 | CD19 | Lenalidomide and Blinatumomab for the Treatment of Relapsed Non-Hodgkin Lymphoma | Not yet recruiting | Blinatumumab + lenalidomide |
| NCT number | Multispecific T-cell engager | Target | Title | Status | Intervention |
|---|---|---|---|---|---|
| NCT04623541 | Epcoritamab | CD20 | Safety and Efficacy Study of Epcoritamab in Subjects With Relapsed/Refractory Chronic Lymphocytic Leukemia and Richter's Syndrome | Recruiting | Monotherapy or combination therapy: epcoritamab + venetoclax epcoritamab + lenalidomide epcoritamab + R-CHOP (ie, rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine (Oncovin), and prednisone) |
| NCT06564038 | AZD0486 | CD19 | A Study of AZD0486 Monotherapy or in Combination With Other Anti-Cancer Agents for Mature B-Cell Malignancies | Not yet recruiting | AZD0486 + acalabrutinib, prednisone, rituximab |
| NCT04763083 | NVG-111 | ROR1 | First in Human Study of NVG-111 in Relapsed/Refractory ROR1+ Malignancies Conditions | Recruiting | Monotherapy |
| NCT00889408 | DT2219ARL | CD19 and CD22 | DT2219ARL for Relapsed or Refractory CD19 (+), CD 22 (+) B-Lineage Leukemia or Lymphoma | Completed | Monotherapy |
| NCT02568553 | CD19 | Lenalidomide and Blinatumomab for the Treatment of Relapsed Non-Hodgkin Lymphoma | Not yet recruiting | Blinatumumab + lenalidomide |
In conclusion, bsAbs and BiTEs represent a promising class of immunotherapeutic agents for CLL. Preclinical studies have shown their potential to induce robust antitumor responses, while early clinical trials have demonstrated their safety and efficacy with repeat dosing.
Limitations of autologous T-cell–based treatments in CLL
Although initial CAR T-cell studies included CLL patients, the efficacy of these therapies has lagged significantly compared to their success in aggressive leukemias and other lymphomas. Similarly, while monotherapy with bsAbs has shown some efficacy, most observed responses have been partial. Low response rates have been attributed to acquired T-cell defects that accumulate upon disease progression (Figure 2). T-cell abnormalities include a skewed distribution and functional impairments, including a reduced capacity to activate, proliferate, and exert cytotoxicity upon T-cell receptor (TCR) or CAR stimulation.31-36 While some of these differences may be related to the natural aging process of the immune system, exacerbated dysfunction is also consistently observed when compared to age-matched healthy individuals.24,32,37 As a consequence of skewing toward effector cells, the increased expression of inhibitory receptors such as PD-1, CD244, LAG-3, and CD160 on T cells from CLL patients have been found. Blocking these receptors in vitro improves T-cell function,38 but clinical studies with immune checkpoint inhibitors have been very disappointing, as remissions were not seen in CLL as they were in patients with RS.39
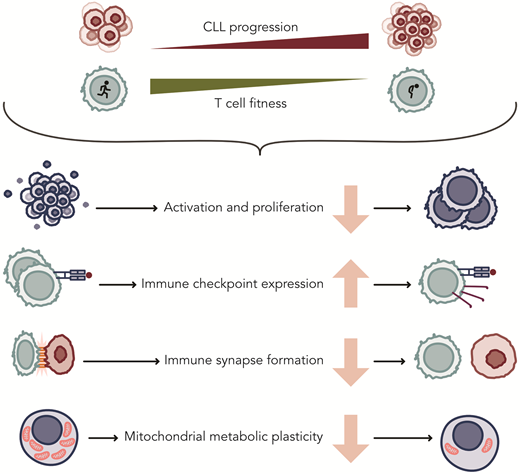
Acquired changes in T cells of patients with CLL that affect their function. Professional ilustration by Somersault18:24.
Acquired changes in T cells of patients with CLL that affect their function. Professional ilustration by Somersault18:24.
One of the earliest indications of defective T-cell activation was the discovery of defective immune synapse formation between CLL cells and T cells, a defect that can also be replicated in healthy donor T cells cocultured with CLL cells.31 Costimulatory and adhesion molecules, critical for proper IS formation and intracellular signaling upon TCR ligation, are poorly expressed by circulating CLL cells compared to normal B cells, contributing to their poor antigen-presenting function.32 Another explanation for poor T-cell activation in CLL patients following TCR ligation is an imbalance between activating and inhibitory signals, potentially involving the phosphatase SIGLEC-10.40 Metabolic plasticity is essential for T-cell function, as cells need to adjust their energy consumption to meet cellular demands and nutrient availability. T cells from CLL patients fail to upregulate glycolysis upon activation, which is necessary for effective activation, differentiation, and proliferation. The PI3K-Akt-mTOR axis, crucial for T-cell survival and metabolic reprogramming, appears dysregulated in CLL,35 contributing to an imbalance favoring effector T cells over naive and memory T cells. This overall T-cell dysfunction might also explain the higher CAR-T manufacturing failure rates of around 20% to 25% in CLL. The exception has been liso-cel, which has a 96% manufacturing success.41-43
Future perspectives
Modifications in CAR design and platforms
Modifications to CAR T-cell constructs to enhance their efficacy in CLL and other diseases where second-generation CAR T cells have shown suboptimal results due to immunocompromised states are actively being explored.44 These modifications aim to improve T-cell expansion, cytokine production, persistence, and antitumor activity by incorporating additional costimulatory domains or transgenes. Third-generation CAR T cells, which include multiple costimulatory domains like CD28 and 4-1BB, have shown promising safety profiles in preliminary studies, but efficacy data in CLL remain limited.45,46 Fourth-generation CAR T cells, also known as TRUCKs, incorporate additional transgenes to enhance effector functions or counteract immunosuppressive signals from the tumor microenvironment. Early results from a study using CD28-costimulated CAR T cells expressing 4-1BB- ligand reported encouraging CR rates in CLL and RS patients.47 Early work in direct vector injection/infusion into patients is just beginning, and this could produce in vivo CAR T cells quickly, thereby saving time, money and resources.48
Combining autologous T-cell–based strategies with targeted agents
Ibrutinib, a BTKi with antitumor and immunomodulatory effects, has been shown to improve CAR T-cell expansion, phenotype, and functionality when administered before or concurrently with CAR T-cell therapy. A phase 1/2 trial combining JCAR014 with ibrutinib in relapsed/refractory CLL patients reported an 83% ORR, with 22% CR,18 while another study using CTL119 with ibrutinib achieved a 69% ORR and a 44% CR rate.49 While these results are promising, it remains unclear whether the combination of CAR T and ibrutinib provides a significant overall improvement in efficacy compared to CAR-T therapy alone. The potential benefits of adding ibrutinib must be carefully weighed against its associated side effects.50 The ongoing TRANSCEND-CLL-004 trial includes an ibrutinib-lisocel cohort, and its longer-term results will be of significant interest to the field.
Other potential combination strategies include PD-1/PD-L1 inhibitors and lenalidomide, which have shown promise in enhancing T-cell function in preclinical models,31,38,51 though clinical data in CLL are pending.
Additionally, the incorporation of PI3K inhibitors (such as idelalisib and duvelisib) into the CAR-T manufacturing process shows promise for enhancing the final product.52 When used ex vivo during manufacturing, an increase in the frequency of naive and central memory T cells and the enhancement of cytotoxic potential occurs. Also, ex vivo coculture with these agents improves CAR T-cell expansion and persistence without the systemic toxicities associated with in vivo administration.53,54 Based on preclinical data, the optimal timing for PI3K inhibitor administration is during the ex vivo expansion phase, allowing the modulation of T-cell differentiation and function in a controlled environment.
The in vivo use of PI3K inhibitors poses significant risks due to their effects on T cells and other immune cells. While some studies have explored short courses of PI3K inhibition post CAR-T infusion, the benefits must be carefully weighed against potential toxicities.55 Currently, limiting PI3K inhibitor use to ex vivo manufacturing appears to be the safest and most effective strategy for optimizing CAR-T therapy.
Furthermore, the use of bromodomain and extraterminal inhibitors during CAR T-cell production has led to improved phenotypical characteristics and enhanced metabolic fitness.56 These combination approaches aim to overcome CLL's immunosuppressive nature and improve (CAR) T-cell efficacy.
Conclusions
The treatment landscape for CLL has evolved significantly, with targeted therapies largely replacing CIT. However, resistance or intolerance to these drugs leaves limited options for patients. CAR T-cell therapy has shown promise as a single infusion treatment, but initial clinical trial results have been disappointing, highlighting the need to better understand the factors affecting treatment success. A significant challenge in CLL treatment is the acquired T-cell dysfunction in patients, and understanding its mechanism is crucial for overcoming this issue and improving outcomes. Current research focuses on designing enhanced CAR constructs and platforms, incorporating immunomodulatory compounds into the manufacturing process, and combining targeted treatments with CAR T-cell therapy and/or bsAbs for a consolidation strategy.
Acknowledgment
Arnon P. Kater is supported by ERC Consolidator: BOOTCAMP (864815) and Lymph and Co: 2018-LYCo008.
Conflict-of-interest disclosure
Arnon P. Kater: advisory board/speaker: AbbVie, AstraZeneca, Bristol Myers Squibb, Janssen, LAVA, Roche/Genentech; travel grants: AbbVie, Janssen; research funding: AstraZeneca, Janssen, Roche/Genentech, AbbVie, Bristol Myers Squibb.
Tanya Siddiqi: advisory board/speaker: Astra Zeneca, BeiGene; Bristol Myers Squibb, Celgene, AbbVie, Pharmacyclics, Gilead, Dava Oncology, ResearchToPractice.
Off-label drug use
Arnon P. Kater: Lisocabtagene-maraleucel has label in the United States (not in Europe); other drugs currently off-label: tisagenlecleucel, blinatumomab, epcoritamab.
Tanya Siddiqi: Lisocabtagene-maraleucel has label in the United States (not in Europe); other drugs currently off-label: tisagenlecleucel, blinatumomab, epcoritamab.
