

Abstract
Kidney disease is a common complication of monoclonal immunoglobulin (MIg)–secreting B-cell disorders and predominantly occurs in patients who do not meet the criteria for an overt hematological disease. To distinguish this situation from monoclonal gammopathy of undetermined significance, which lacks organ damage, the term monoclonal gammopathy of renal significance (MGRS) was introduced to depict the association of a small, otherwise indolent B-cell clone, with renal disease induced by the secreted MIg. The spectrum of renal disorders in MGRS is wide, encompassing both tubular and glomerular disorders, classified according to the composition of deposits and their ultrastructural pattern of organization. Renal lesions, independent of the tumor burden, are mostly governed by the molecular characteristics of the MIg variable domain and involve either direct (deposition or precipitation) or indirect (autoantibody activity, complement activation) mechanisms. The diagnosis, often suggested by careful analysis of renal and extrarenal symptoms, almost always requires histological confirmation by a kidney biopsy with light, immunofluorescence, and electron microscopy studies. Most patients do not have a known monoclonal gammopathy at presentation. Hematologic investigations should include serum and urine protein electrophoresis and immunofixation, serum-free light chain measurements, and bone marrow studies with flow cytometry and cytogenetics to determine the nature of the pathogenic clone (most commonly plasmocytic). Early diagnosis before the development of severe chronic kidney disease and rapid achievement of deep hematological response through clone-targeted chemotherapy (currently based on proteasome inhibitor and monoclonal anti-CD38 antibody–based combinations for plasma cell clones) are the main factors influencing long-term renal and patient outcomes.
Learning Objectives
Recognize the different types of renal disorders associated with monoclonal immunoglobulins
Diagnose MGRS
Describe the principles of hematologic and renal management of MGRS
CLINICAL CASE
A 67-year-old man with a history of hypertension and hypercholesterolemia was referred for chronic kidney disease (CKD) fortuitously discovered 4 months ago. On admission, clinical examination showed high blood pressure (155/95 mmHg), bilateral lower limb edema, and hepatomegaly. He denied any alcohol abuse.
Blood tests results were as follows: serum creatinine, 1.6 mg/dL; estimated glomerular filtration rate (eGFR; CKD-EPI equation), 43 mL/min/1.73 m2; total proteins, 8.1 g/dL; albumin, 4.3 g/dL; calcium, 9.6 mg/dL; elevated gamma glutamyl transpeptidase ( × 6 upper limit of normal) and alkaline phosphatase ( × 3 upper limit of normal) levels; and normal aminotransferases. The complete blood count was normal. Urinalysis revealed microscopic hematuria and proteinuria (2.1 g/d, 253 mg/mmol). Tests for anti-DNA, antineutrophil cytoplasmic, antiphospholipase A2 receptor (PLA2R) antibodies, hepatitis B, hepatitis C, and HIV were negative. Serum complement C3 and C4 levels were normal. An abdominal ultrasound scan displayed normal-sized kidneys and hepatomegaly without splenomegaly.
Monoclonal immunoglobulin testing revealed a serum immunoglobulin (IgG kappa) monoclonal gammopathy (1.3 g/dL). Kappa and lambda serum free light chain (FLC) levels were 21.3 mg/dL and 1.1 mg/dL, respectively. Proteinuria was composed of predominant albuminuria (72%) with small kappa Bence Jones proteinuria. Bone marrow flow cytometry analysis showed 4% infiltration of clonal kappa LC-positive plasma cells. next-generation sequencing (NGS)–based cytogenetics revealed the presence of t(11;14) without t(4;14) or del(17p).
The patient was hospitalized for a kidney biopsy.
Overview and pathophysiology of monoclonal gammopathy–associated renal diseases
Among the variety of organ lesions induced by monoclonal immunoglobulins (MIgs), kidney involvement is predominant. The main cause of severe acute kidney injury in multiple myeloma is LC cast nephropathy, which complicates high tumor mass disease, secreting massive amounts of monoclonal LCs that precipitate with uromodulin in distal tubule lumens.1 However, other MIg-related renal diseases occur, usually in the setting of low-grade B-cell clones. Given the absence of criteria for a symptomatic hematologic disorder, the decision to introduce chemotherapy remained challenging until the introduction of monoclonal gammopathy of renal significance (MGRS) in 2012.2 MGRS defines a monoclonal gammopathy produced by a small, otherwise asymptomatic clone, with renal disease induced by the secreted MIg.2,3 The concept was later extended to monoclonal gammopathy of clinical significance (MGCS), covering the full spectrum of organ damage related to MIg by mechanisms other than the tumor burden.4 By establishing a clear distinction from monoclonal gammopathy of undetermined significance (MGUS) devoid of organ damage, these concepts contributed to improving the management and prognosis of MIg-related conditions.
Not all MIg are nephrotoxic. The prevalence of MGRS renal lesions on kidney biopsies from patients with CKD and MGUS is around 40%.5 Nephrotoxicity is an intrinsic property, mostly dictated by the variable domain molecular characteristics (charge, hydrophobicity, glycosylation) induced by somatic hypermutations, which promote self-aggregation and determine tissue interaction and organization of deposits. Although the specific toxicity of a given MIg cannot be predicted by sequence peculiarities, several associations are known. Vλ6 LCs (IGLV6-57 gene) are overrepresented in renal LC-related (AL) amyloidosis, whereas light chain deposition disease (LCDD) predominantly involves Vκ4 LCs showing N-glycosylation sites and cationic complementarity-determining regions favoring their deposition to anionic basement membranes. Fanconi syndrome, defined by generalized proximal tubule dysfunction, is associated with Vκ1 LCs bearing somatic hypermutations in complementarity- determining regions, inducing resistance to proteolysis and crystallization in the lysosomes of proximal tubular cells. MIg heavy chains in heavy chain deposition disease (HCDD) always feature deletion of the first constant domain, required for their secretion as free fragments by plasma cells.6
The distribution of kidney lesions is also influenced by MIg size. Monoclonal LCs, freely filtered through the glomerulus and physiologically reabsorbed by proximal tubule cells, can affect all kidney compartments, whereas entire MIgs that do not cross the glomerular barrier induce glomerular lesions only. Kidney injury most often results from the deposition or precipitation of an entire MIg or a free LC and, exceptionally, a free heavy chain. MIg may deposit as organized substructures or as amorphous aggregates.6 Other mechanisms have been described, including antibody activity toward glomerular antigens (PLA2R, type IV collagen),7-8 leading to immune-complex deposition and complement activation. Toxicity may occur without deposition, as in monoclonal gammopathy–associated thrombotic microangiopathy (TMA) and glomerulonephritis with isolated C3 deposits (C3GN),9,10 resulting from MIg-induced overactivation of the complement alternative pathway, directly or through autoantibody activity against complement regulatory proteins.11
Classification of MGRS-related renal disorders
The International Kidney and Monoclonal Gammopathy Research Group classification distinguishes 3 categories based on the composition of deposits/inclusions and their ultrastructural pattern by electron microscopy (EM) (Figure 1)3:
Nephropathies with organized MIg deposits, including the following: fibrils in AL, heavy chain (AH), and light and heavy chain (AHL)-related amyloidosis12; microtubules in immunotactoid glomerulopathy (ITGP) and type 1 and 2 cryoglobulinemic glomerulonephritis13-15; crystals in crystalglobulinemia and cryocrystal-cryoglobulinemia, LC crystalline podocytopathy, LC-associated Fanconi syndrome, and crystal-storing histiocytosis (CSH) (Table 1 and Figure 2).16-20
Nephropathies with amorphous (nonorganized) MIg deposits featuring monoclonal immunoglobulin deposition disease (subdivided into LCDD, HCDD, and light and heavy chain [LHCDD] deposition disease) and proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) (Table 2 and Figure 3A-B).21-26
Nephropathies without MIg deposits: monoclonal gammopathy–associated renal TMA and C3 glomerulopathy (C3GP), which includes C3GN and dense deposit disease (Table 2 and Figure 3B).9-11
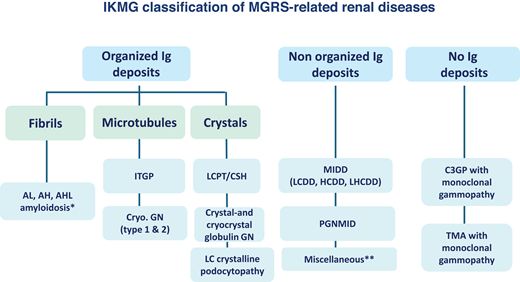
International Kidney and Monoclonal Gammopathy Research Group classification of MGRS-related renal diseases. *Fibrillary glomerulonephritis (FGN) is a glomerular disorder characterized by immunoglobulin deposits composed of IgG1 and IgG4 and organized into randomly oriented fibrils of 9 to 26 nm in diameter at the ultrastructural level. These deposits coexist with abundant glomerular expression of DNAJB9, which is a sensitive and specific marker of the disease. Although the vast majority of DNAJB9-positive FGN cases feature polyclonal IgG deposits, restriction for either kappa or lambda light chain is observed by conventional immunofluorescence in around 10% of cases. FGN was initially classified as an MGRS-related renal disorder. However, a 2020 series from the Mayo Clinic in which kidney biopsies were reviewed using paraffin immunofluorescence and IgG subclass staining clearly established that DNAJB9-positive monotypic FGN is very rare and, with few exceptions, is not associated with monoclonal gammopathy.28 Thus, monotypic FGN was excluded from the 2021 International Kidney and Monoclonal Gammopathy Research Group classification of MGRS-related renal disorders.3 **Examples include anti-GBM disease secondary to a monoclonal gammopathy and membranous nephropathy with monoclonal IgG deposits (with or without anti-PLA2R autoantibody activity). Cryo.GN, cryoglobulinemic glomerulonephritis; GBM, glomerular basement membrane; LCPT, light chain proximal tubulopathy. Adapted with permission from Leung et al.3
International Kidney and Monoclonal Gammopathy Research Group classification of MGRS-related renal diseases. *Fibrillary glomerulonephritis (FGN) is a glomerular disorder characterized by immunoglobulin deposits composed of IgG1 and IgG4 and organized into randomly oriented fibrils of 9 to 26 nm in diameter at the ultrastructural level. These deposits coexist with abundant glomerular expression of DNAJB9, which is a sensitive and specific marker of the disease. Although the vast majority of DNAJB9-positive FGN cases feature polyclonal IgG deposits, restriction for either kappa or lambda light chain is observed by conventional immunofluorescence in around 10% of cases. FGN was initially classified as an MGRS-related renal disorder. However, a 2020 series from the Mayo Clinic in which kidney biopsies were reviewed using paraffin immunofluorescence and IgG subclass staining clearly established that DNAJB9-positive monotypic FGN is very rare and, with few exceptions, is not associated with monoclonal gammopathy.28 Thus, monotypic FGN was excluded from the 2021 International Kidney and Monoclonal Gammopathy Research Group classification of MGRS-related renal disorders.3 **Examples include anti-GBM disease secondary to a monoclonal gammopathy and membranous nephropathy with monoclonal IgG deposits (with or without anti-PLA2R autoantibody activity). Cryo.GN, cryoglobulinemic glomerulonephritis; GBM, glomerular basement membrane; LCPT, light chain proximal tubulopathy. Adapted with permission from Leung et al.3
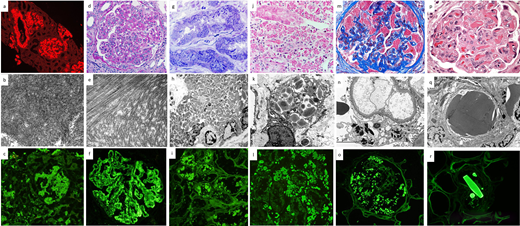
Pathology of MGRS lesions with organized Ig deposits. (a-c): AL amyloidosis: review of Congo red stain under the Texas red immunofluorescence filter reveals bright red smudgy deposits in glomeruli, vessels, and focally in the interstitium (a, × 100). The deposits on transmission EM are composed of randomly oriented straight fibrils (b, × 40 000). On immunofluorescence, smudgy amyloid deposits in glomeruli and interstitium stain positive for lambda light chain (c, × 200) but not for kappa light chain (not shown). (d-f): Immunotactoid glomerulopathy. The glomerulus exhibits global mesangial and endocapillary hypercellularity with intracapillary infiltrating lymphocytes and monocytes (d, PAS, × 400). The deposits on EM are composed of microtubular structures with parallel alignment (e, × 40 000). On immunofluorescence, there is bright granular to semilinear glomerular positivity for IgG (f, × 400) and kappa light chain but not for lambda light chain (not shown). (g-i): Light chain proximal tubulopathy. Numerous osmophilic crystals with needle and rod shapes are seen within proximal tubular cells (g, toluidine blue–stained EM survey section, × 400). EM reveals the engorgement of proximal tubular cells by rhomboidal and rod-shaped crystals, causing compression of the nuclear contours (h, × 4400). Immunofluorescence on paraffin tissue after antigen retrieval with pronase reveals staining of proximal tubular cells for kappa light chain (i, × 400) but not for lambda light chain (not shown). (j-l): Light chain CSH. There is interstitial infiltration of histiocytes containing intracytoplasmic red crystalline inclusions (j, trichrome stain, × 400). A high-power EM image shows rhomboidal and rod-shaped intralysosomal crystals in an interstitial histiocyte (k, × 8000). Immunofluorescence on paraffin tissue after antigen retrieval with pronase reveals staining of crystals within interstitial histiocytes for kappa light chain (l, ×400) but not for lambda light chain (not shown). (m-o): Light chain crystalline podocytopathy. Podocytes are filled with trichrome-red crystalline inclusions. There is partial collapse of the underlying glomerular tuft (m, trichrome stain, × 600). High-power EM image shows highly electron dense crystals within podocytes (n, × 8000). Many rod-shaped and rhomboidal inclusions within podocytes stain positive for kappa light chain by immunofluorescence on paraffin tissue after antigen retrieval with pronase (o, × 400) but not for lambda light chain (not shown). (p-r): Crystalglobulin-induced nephropathy. Many eosinophilic, needle-shaped crystals are present within glomerular capillaries, associated with a macrophage-rich inflammatory reaction and mild mesangiolysis (p, hematoxylin and eosin, × 600). Large highly electron-dense extracellular crystals are seen plugging the glomerular capillaries. Similar but fewer crystals are also seen in the subendothelial region and mesangium (q, EM, × 8000). Large crystals in vessels stain brightly for IgG by immunofluorescence on paraffin tissue after antigen retrieval with pronase (r, × 400) and kappa light chain but not for lambda light chain (not shown).
Pathology of MGRS lesions with organized Ig deposits. (a-c): AL amyloidosis: review of Congo red stain under the Texas red immunofluorescence filter reveals bright red smudgy deposits in glomeruli, vessels, and focally in the interstitium (a, × 100). The deposits on transmission EM are composed of randomly oriented straight fibrils (b, × 40 000). On immunofluorescence, smudgy amyloid deposits in glomeruli and interstitium stain positive for lambda light chain (c, × 200) but not for kappa light chain (not shown). (d-f): Immunotactoid glomerulopathy. The glomerulus exhibits global mesangial and endocapillary hypercellularity with intracapillary infiltrating lymphocytes and monocytes (d, PAS, × 400). The deposits on EM are composed of microtubular structures with parallel alignment (e, × 40 000). On immunofluorescence, there is bright granular to semilinear glomerular positivity for IgG (f, × 400) and kappa light chain but not for lambda light chain (not shown). (g-i): Light chain proximal tubulopathy. Numerous osmophilic crystals with needle and rod shapes are seen within proximal tubular cells (g, toluidine blue–stained EM survey section, × 400). EM reveals the engorgement of proximal tubular cells by rhomboidal and rod-shaped crystals, causing compression of the nuclear contours (h, × 4400). Immunofluorescence on paraffin tissue after antigen retrieval with pronase reveals staining of proximal tubular cells for kappa light chain (i, × 400) but not for lambda light chain (not shown). (j-l): Light chain CSH. There is interstitial infiltration of histiocytes containing intracytoplasmic red crystalline inclusions (j, trichrome stain, × 400). A high-power EM image shows rhomboidal and rod-shaped intralysosomal crystals in an interstitial histiocyte (k, × 8000). Immunofluorescence on paraffin tissue after antigen retrieval with pronase reveals staining of crystals within interstitial histiocytes for kappa light chain (l, ×400) but not for lambda light chain (not shown). (m-o): Light chain crystalline podocytopathy. Podocytes are filled with trichrome-red crystalline inclusions. There is partial collapse of the underlying glomerular tuft (m, trichrome stain, × 600). High-power EM image shows highly electron dense crystals within podocytes (n, × 8000). Many rod-shaped and rhomboidal inclusions within podocytes stain positive for kappa light chain by immunofluorescence on paraffin tissue after antigen retrieval with pronase (o, × 400) but not for lambda light chain (not shown). (p-r): Crystalglobulin-induced nephropathy. Many eosinophilic, needle-shaped crystals are present within glomerular capillaries, associated with a macrophage-rich inflammatory reaction and mild mesangiolysis (p, hematoxylin and eosin, × 600). Large highly electron-dense extracellular crystals are seen plugging the glomerular capillaries. Similar but fewer crystals are also seen in the subendothelial region and mesangium (q, EM, × 8000). Large crystals in vessels stain brightly for IgG by immunofluorescence on paraffin tissue after antigen retrieval with pronase (r, × 400) and kappa light chain but not for lambda light chain (not shown).
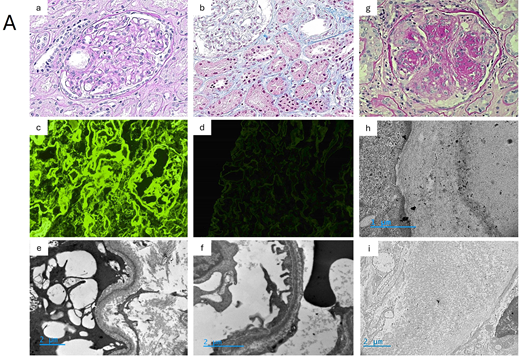
(A) Pathology of MGRS lesions with nonorganized deposits: MIDD. (a-f): Patient's kidney biopsy, light chain deposition disease. Glomeruli show mild mesangial sclerosis, with thickened capillary walls and Bowman's capsule membrane (a, PAS stain, × 400). Mildly thickened tubular basement membranes show Congo red negative deposits (not shown) (b, trichrome stain, × 400). On immunofluorescence, diffuse linear deposits of kappa light chains are seen in the mesangium, around capillary walls, and in the tubular basement membranes (c, × 400), whereas no staining is observed for lambda light chain (d, × 400). On EM, linear “powdery punctuate” deposits are visible on the outer aspect of tubular basement membranes (e, × 10 000) and on the inner aspect of glomerular basement membranes (f, × 15 000). LCDD: typical pattern of nodular glomerulosclerosis with aneurysmal dilatation of glomerular capillary loops (g, PAS stain, × 400). LCDD: EM showing linear glomerular deposits stained with a gold conjugated anti-kappa light chain antibody (h, × 30 000), without significant staining for lambda light chain (i, × 10 000).
(A) Pathology of MGRS lesions with nonorganized deposits: MIDD. (a-f): Patient's kidney biopsy, light chain deposition disease. Glomeruli show mild mesangial sclerosis, with thickened capillary walls and Bowman's capsule membrane (a, PAS stain, × 400). Mildly thickened tubular basement membranes show Congo red negative deposits (not shown) (b, trichrome stain, × 400). On immunofluorescence, diffuse linear deposits of kappa light chains are seen in the mesangium, around capillary walls, and in the tubular basement membranes (c, × 400), whereas no staining is observed for lambda light chain (d, × 400). On EM, linear “powdery punctuate” deposits are visible on the outer aspect of tubular basement membranes (e, × 10 000) and on the inner aspect of glomerular basement membranes (f, × 15 000). LCDD: typical pattern of nodular glomerulosclerosis with aneurysmal dilatation of glomerular capillary loops (g, PAS stain, × 400). LCDD: EM showing linear glomerular deposits stained with a gold conjugated anti-kappa light chain antibody (h, × 30 000), without significant staining for lambda light chain (i, × 10 000).
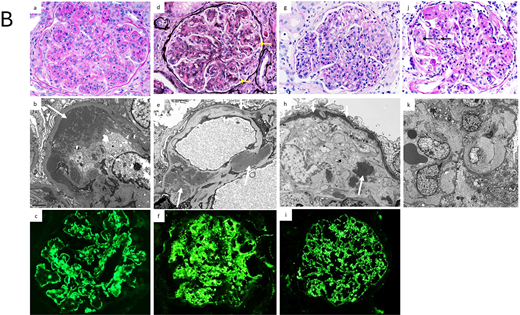
(B) Pathology of other MGRS lesions with nonorganized deposits and MGRS lesions without Ig deposits. (a-c): Proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Glomeruli exhibit lobular accentuation due to prominent mesangial expansion by mesangial hypercellularity and sclerosis, with associated global duplication of the glomerular basement membranes and cellular interposition (a, PAS stain, × 400). On EM, large granular subendothelial nonorganized electron dense deposits (arrow) are seen (b, × 8000). On immunofluorescence, there is granular to semilinear global glomerular capillary wall and mesangial staining for IgG (c, ×400) and kappa light chain, with negative staining for lambda light chain (not shown). (d-f): C3 glomerulonephritis associated with monoclonal gammopathy. The glomerulus shows global mesangial expansion by an increase in mesangial cell number and the presence of glassy silver-negative immune deposits. There is segmental duplication of the glomerular basement membranes (arrows) (d, silver stain, × 400). On EM, large mesangial (large arrows) and small intramembranous (small arrow) electron dense deposits are evident (e, × 8000). On immunofluorescence, there is bright global granular mesangial and glomerular basement membrane staining for C3 (f, × 400). Glomeruli were negative for immunoglobulins and C1q (not shown). (g-i): Dense deposit disease associated with monoclonal gammopathy. The glomerulus exhibits global endocapillary hypercellularity with numerous intracapillary infiltrating neutrophils with some lymphocytes and monocytes (g, × 400). An EM image showing highly electron dense deposits occupying most of the thickness of the glomerular basement membrane (small arrows), with nodular deposits in the mesangium (large arrow) (h, × 6000). On immunofluorescence, there is bright global granular glomerular basement membrane and mesangial staining for C3 (i, × 400). Glomeruli were negative for immunoglobulins and C1q (not shown). (j-k): Glomerular microangiopathy associated with monoclonal gammopathy. The glomerulus exhibits segmental mesangiolysis (arrows) and duplication of the glomerular basement membrane (j, × 400). On EM images from a patient with POEMS syndrome shows global mesangiolysis and marked widening of the subendothelial zone by electron lucent fluffy material (k, × 4000). POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes.
(B) Pathology of other MGRS lesions with nonorganized deposits and MGRS lesions without Ig deposits. (a-c): Proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Glomeruli exhibit lobular accentuation due to prominent mesangial expansion by mesangial hypercellularity and sclerosis, with associated global duplication of the glomerular basement membranes and cellular interposition (a, PAS stain, × 400). On EM, large granular subendothelial nonorganized electron dense deposits (arrow) are seen (b, × 8000). On immunofluorescence, there is granular to semilinear global glomerular capillary wall and mesangial staining for IgG (c, ×400) and kappa light chain, with negative staining for lambda light chain (not shown). (d-f): C3 glomerulonephritis associated with monoclonal gammopathy. The glomerulus shows global mesangial expansion by an increase in mesangial cell number and the presence of glassy silver-negative immune deposits. There is segmental duplication of the glomerular basement membranes (arrows) (d, silver stain, × 400). On EM, large mesangial (large arrows) and small intramembranous (small arrow) electron dense deposits are evident (e, × 8000). On immunofluorescence, there is bright global granular mesangial and glomerular basement membrane staining for C3 (f, × 400). Glomeruli were negative for immunoglobulins and C1q (not shown). (g-i): Dense deposit disease associated with monoclonal gammopathy. The glomerulus exhibits global endocapillary hypercellularity with numerous intracapillary infiltrating neutrophils with some lymphocytes and monocytes (g, × 400). An EM image showing highly electron dense deposits occupying most of the thickness of the glomerular basement membrane (small arrows), with nodular deposits in the mesangium (large arrow) (h, × 6000). On immunofluorescence, there is bright global granular glomerular basement membrane and mesangial staining for C3 (i, × 400). Glomeruli were negative for immunoglobulins and C1q (not shown). (j-k): Glomerular microangiopathy associated with monoclonal gammopathy. The glomerulus exhibits segmental mesangiolysis (arrows) and duplication of the glomerular basement membrane (j, × 400). On EM images from a patient with POEMS syndrome shows global mesangiolysis and marked widening of the subendothelial zone by electron lucent fluffy material (k, × 4000). POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes.
Characteristics of the main MGRS-related renal disorders with organized Ig deposits
| Disease | Renal pathology | Renal manifestations | Extrarenal manifestations | Hematologic findings | Hematologic diagnosis |
|---|---|---|---|---|---|
| Immunoglobulinic amyloidosis: AL amyloidosis (94%) AH/AHL amyloidosis (6%) | LM: Congo red positive glomerular (pred. mesangial), vascular +/− interstitial deposits IF (composition of deposits): AL: LC-only deposits (mostly λ) AH: HC-only deposits (mostly γ1 or γ4) with CH1 deletion AHL: monotypic LC and HC (mostly γ) deposits EM: Nonbranched, randomly arranged fibrils (7-12 nm) | CKD (median s. creat. 1.2 mg/dL) Massive proteinuria (median 5-6 g/d) Nephrotic syndrome (>60%) Hematuria/hypertension rare | Frequent: Cardiomyopathy (70%) Liver disease GI tract Peripheral neuropathy Autonomic dysfunction Carpal tunnel Soft tissues Macroglossia Periorbital hematoma | Positive serum/urine EP/IFix: 80% Abnormal sFLC level: 90% Plasma cell clone: 90% (t(11;14): 50%) Lymphocytic/LPC clone: 10% (IgM secreting) Factor X deficiency: 10% Hypocomplementemia: no | MGCS/MGRS: 80% MM: 16% WM, B-cell lymphoma: 4% |
| Immunotactoid glomerulopathy | LM: MPGN, EPGN, MGN IF: monotypic IgG (mostly IgG1κ) granular deposits in mesangium and CW EM: parallel microtubules (14-60 nm) | CKD (median s. creat 1.5 mg/dL) Heavy proteinuria (median 6 g/d) Nephrotic syndrome (~70%) Hematuria (75%-90%) Hypertension (60%-80%) | Extremely rare: Skin capillaritis Mononeuritis multiplex | Positive serum/urine EP/IFix: 50% Abnormal sFLC level: 20%-30% Plasma cell clone: 50% Lymphocytic clone: 50% Hypocomplementemia: 33% | MGRS: 50% CLL: 40%-45% B-cell lymphoma: 10% MM: 4% |
| Type 1 cryoglobulinic GN | LM: MPGN, EPGN with glomerular monocyte infiltration, frequent intracapillary immune thrombi IF: monotypic Ig (mostly IgGκ or IgMκ) granular glomerular +/− vascular deposits EM: microtubular and/or crystalline deposits (extra +/− intracellular) if IgG or IgA | CKD (median s. creat. 3 mg/dL) Heavy proteinuria (median 3 g/d) Nephrotic syndrome (40%), Hematuria/ hypertension (70%) | Frequent (60%): Pupura, skin necrosis/gangrene, livedo reticularis Raynaud phenomenon Peripheral neuropathy Arthralgias/arthritis GI tract | Positive serum/urine EP/IFix: 80% Abnormal sFLC level: 40% Plasma cell clone: 40% Lymphocytic/LPC clone: 60% Type 1 cryoglobulinemia: 60% Hypocomplementemia (mostly low C4) (60%) | MGRS: 80% CLL: 10% WM/ lymphoma: 10%-50% MM: 3%-7% |
| LC crystalline podocytopathy | LM: FSGS (60%) IF: LC-only crystals (κ 90%) EM: intracytoplasmic crystals in podocytes | CKD (median s. creat. 1.9 mg/dL) Proteinuria (median 3.4 g/d) Nephrotic syndrome (30%) | Common: LC keratopathy (25%) | Positive serum/urine EP/IFix: 100% Abnormal sFLC level: 80% Plasma cell clone: ~100% Lymphocytic clone: anecdotal | MGRS: 55% MM: 45% |
| LC proximal tubulopathy | LM: PTC swelling, dedifferentiation IF: PTC LC staining (mostly κ in crystalline variant), rarely λ (noncrystalline variant) EM: PTC LC crystals (if κ) or lysosomal inclusions | CKD (median s.creat 2 mg/dL) Proteinuria (median 1.5-2.5 g/d) Proximal tubulopathy with or without complete FS | Common: If FS: osteomalacia, stress fractures IF CSH: lungs, GI tract, cornea, bone marrow, lymph nodes, liver, spleen | Positive serum/urine PEP/IFix: ~100% Abnormal sFLC level: ~100% Plasma cell clone: 95% Lymphocytic/LPC clone: 5% | MGRS: 60%-80% MM: 15%-30% WM/ lymphoma: 3%-8% |
| Disease | Renal pathology | Renal manifestations | Extrarenal manifestations | Hematologic findings | Hematologic diagnosis |
|---|---|---|---|---|---|
| Immunoglobulinic amyloidosis: AL amyloidosis (94%) AH/AHL amyloidosis (6%) | LM: Congo red positive glomerular (pred. mesangial), vascular +/− interstitial deposits IF (composition of deposits): AL: LC-only deposits (mostly λ) AH: HC-only deposits (mostly γ1 or γ4) with CH1 deletion AHL: monotypic LC and HC (mostly γ) deposits EM: Nonbranched, randomly arranged fibrils (7-12 nm) | CKD (median s. creat. 1.2 mg/dL) Massive proteinuria (median 5-6 g/d) Nephrotic syndrome (>60%) Hematuria/hypertension rare | Frequent: Cardiomyopathy (70%) Liver disease GI tract Peripheral neuropathy Autonomic dysfunction Carpal tunnel Soft tissues Macroglossia Periorbital hematoma | Positive serum/urine EP/IFix: 80% Abnormal sFLC level: 90% Plasma cell clone: 90% (t(11;14): 50%) Lymphocytic/LPC clone: 10% (IgM secreting) Factor X deficiency: 10% Hypocomplementemia: no | MGCS/MGRS: 80% MM: 16% WM, B-cell lymphoma: 4% |
| Immunotactoid glomerulopathy | LM: MPGN, EPGN, MGN IF: monotypic IgG (mostly IgG1κ) granular deposits in mesangium and CW EM: parallel microtubules (14-60 nm) | CKD (median s. creat 1.5 mg/dL) Heavy proteinuria (median 6 g/d) Nephrotic syndrome (~70%) Hematuria (75%-90%) Hypertension (60%-80%) | Extremely rare: Skin capillaritis Mononeuritis multiplex | Positive serum/urine EP/IFix: 50% Abnormal sFLC level: 20%-30% Plasma cell clone: 50% Lymphocytic clone: 50% Hypocomplementemia: 33% | MGRS: 50% CLL: 40%-45% B-cell lymphoma: 10% MM: 4% |
| Type 1 cryoglobulinic GN | LM: MPGN, EPGN with glomerular monocyte infiltration, frequent intracapillary immune thrombi IF: monotypic Ig (mostly IgGκ or IgMκ) granular glomerular +/− vascular deposits EM: microtubular and/or crystalline deposits (extra +/− intracellular) if IgG or IgA | CKD (median s. creat. 3 mg/dL) Heavy proteinuria (median 3 g/d) Nephrotic syndrome (40%), Hematuria/ hypertension (70%) | Frequent (60%): Pupura, skin necrosis/gangrene, livedo reticularis Raynaud phenomenon Peripheral neuropathy Arthralgias/arthritis GI tract | Positive serum/urine EP/IFix: 80% Abnormal sFLC level: 40% Plasma cell clone: 40% Lymphocytic/LPC clone: 60% Type 1 cryoglobulinemia: 60% Hypocomplementemia (mostly low C4) (60%) | MGRS: 80% CLL: 10% WM/ lymphoma: 10%-50% MM: 3%-7% |
| LC crystalline podocytopathy | LM: FSGS (60%) IF: LC-only crystals (κ 90%) EM: intracytoplasmic crystals in podocytes | CKD (median s. creat. 1.9 mg/dL) Proteinuria (median 3.4 g/d) Nephrotic syndrome (30%) | Common: LC keratopathy (25%) | Positive serum/urine EP/IFix: 100% Abnormal sFLC level: 80% Plasma cell clone: ~100% Lymphocytic clone: anecdotal | MGRS: 55% MM: 45% |
| LC proximal tubulopathy | LM: PTC swelling, dedifferentiation IF: PTC LC staining (mostly κ in crystalline variant), rarely λ (noncrystalline variant) EM: PTC LC crystals (if κ) or lysosomal inclusions | CKD (median s.creat 2 mg/dL) Proteinuria (median 1.5-2.5 g/d) Proximal tubulopathy with or without complete FS | Common: If FS: osteomalacia, stress fractures IF CSH: lungs, GI tract, cornea, bone marrow, lymph nodes, liver, spleen | Positive serum/urine PEP/IFix: ~100% Abnormal sFLC level: ~100% Plasma cell clone: 95% Lymphocytic/LPC clone: 5% | MGRS: 60%-80% MM: 15%-30% WM/ lymphoma: 3%-8% |
CH1, first constant domain; CW, glomerular capillary wall; EP, electrophoresis; EPGN, endocapillary proliferative glomerulonephritis; FS, renal Fanconi syndrome; FSGS, focal segmental glomerulosclerosis; GI, gastrointestinal; GN, glomerulonephritis; IF, immunofluorescence; IFix, immunofixation; LM, light microscopy; LPC, lymphoplasmacytic; MGN; membranous glomerulopathy; MM, multiple myeloma; MPGN, membranoproliferative glomerulonephritis; PTC, proximal tubular cells; s.creat, serum creatinine; sFLC, serum free light chains; WM, Waldenström macroglobulinemia.
Characteristics of MGRS-related renal disorders with nonorganized Ig deposits and without Ig deposits
| Disease | Renal pathology | Renal manifestations | Extrarenal manifestations | Hematologic findings | Hematologic diagnosis |
|---|---|---|---|---|---|
| MGRS-related renal disorders with nonorganized Ig deposits | |||||
| Monoclonal immunoglobulin deposition disease | LM: nodular glomerulosclerosis (2/3 of cases) with thickened TBM IF: linear deposits along GBM, TBM, and vessels LCDD: LC-only deposits (mostly κ) HCDD: HC-only deposits with CH1 deletion (mostly γ) LHCDD: deposits containing 1 LC and 1 HC lacking CH1 (mostly γ) EM: punctate linear amorphous deposits along GBM, TBM, vessels | CKD (median s.creat. 3 mg/dL) Proteinuria (median 2 g/d) Nephrotic syndrome (22%), Hematuria (60%) Hypertension (60%-80%) | Common in LCDD (35%): Cardiac disease Liver disease Peripheral neuropathy | Positive serum/urine EP/IFix: 90% Abnormal sFLC level: 99%-100% Plasma cell clone: 95%-100% Lymphocytic/LPC clone: <5% Hypocomplementemia (low C4) if HCDDγ1 or HCDDγ3 | MGRS: 65%-80% MM: 20%-35% WM/B-cell lymphoma: 2% |
| Proliferative glomerulonephritis with monoclonal immunoglobulin deposits | LM: MPGN, EPGN, MGN IF: monotypic IgG (mostly IgG3κ, rarely IgM, IgA, LC only); granular deposits in mesangium and CW EM: discontinuous nonorganized glomerular deposits | CKD (mean s.creat. 2.8 mg/dL) Heavy proteinuria (mean 6 g/d) Nephrotic syndrome (50%) Hematuria (80%) Hypertension (38%) | Absent | Positive serum/urine EP/IFix: 20%-30% Abnormal sFLC: 20% Plasma cell clone: ~10% Lymphocytic/LPC clone: ~10% Hypocomplementemia 20% Negative tests for cryoglobulins | MGRS: 95% MM: 3%-4% WM/B-cell lymphoma: 1% |
| MGRS-related renal disorders without Ig deposits | |||||
| C3GP with monoclonal gammopathy | LM: MPGN, MesPGN, EPGN IF: granular deposits of C3 only in mesangium and CW EM: C3GN: less electron dense subendothelial/subepithelial/ mesangial deposits Dense deposit disease: highly electron dense sausage-like deposits within GBM | CKD (median s.creat.1.8 mg/dL) Proteinuria (median 3 g/d) Nephrotic syndrome (43%), Hematuria (90%) | Rare: Digital ischemia | Positive serum/urine EP/IFix: 100% Abnormal sFLC level: 50%-75% Plasma cell clone: 95% Lymphocytic clone: 5% Low C3 (35%-45%) High serum C5bC9: 80% | MGRS: 80%-90% MM: 5%-15% B-cell lymphoma: 6% |
| Thrombotic microangiopathy with monoclonal gammopathy | LM and EM: GBM duplication, mesangiolysis, subendothelial widening “fluff,” thrombosis IF: no Ig deposits | CKD (median s.creat 3 mg/dL) Proteinuria (median 2-4 g/d) Hematuria (90%) | Common: MAHA (50%-90%) CNS, skin, peripheral neuropathy | Positive serum/urine EP/IFix: 100%; Abnormal sFLC level: 28% Plasma cell clone: 80% Lymphocytic/LPC clone: 20% Low C3 (33%) High serum C5bC9: 77% | MGRS: 90% POEMS syndrome: 10% MM: 5% WM/B-cell lymphoma: 5% |
| Disease | Renal pathology | Renal manifestations | Extrarenal manifestations | Hematologic findings | Hematologic diagnosis |
|---|---|---|---|---|---|
| MGRS-related renal disorders with nonorganized Ig deposits | |||||
| Monoclonal immunoglobulin deposition disease | LM: nodular glomerulosclerosis (2/3 of cases) with thickened TBM IF: linear deposits along GBM, TBM, and vessels LCDD: LC-only deposits (mostly κ) HCDD: HC-only deposits with CH1 deletion (mostly γ) LHCDD: deposits containing 1 LC and 1 HC lacking CH1 (mostly γ) EM: punctate linear amorphous deposits along GBM, TBM, vessels | CKD (median s.creat. 3 mg/dL) Proteinuria (median 2 g/d) Nephrotic syndrome (22%), Hematuria (60%) Hypertension (60%-80%) | Common in LCDD (35%): Cardiac disease Liver disease Peripheral neuropathy | Positive serum/urine EP/IFix: 90% Abnormal sFLC level: 99%-100% Plasma cell clone: 95%-100% Lymphocytic/LPC clone: <5% Hypocomplementemia (low C4) if HCDDγ1 or HCDDγ3 | MGRS: 65%-80% MM: 20%-35% WM/B-cell lymphoma: 2% |
| Proliferative glomerulonephritis with monoclonal immunoglobulin deposits | LM: MPGN, EPGN, MGN IF: monotypic IgG (mostly IgG3κ, rarely IgM, IgA, LC only); granular deposits in mesangium and CW EM: discontinuous nonorganized glomerular deposits | CKD (mean s.creat. 2.8 mg/dL) Heavy proteinuria (mean 6 g/d) Nephrotic syndrome (50%) Hematuria (80%) Hypertension (38%) | Absent | Positive serum/urine EP/IFix: 20%-30% Abnormal sFLC: 20% Plasma cell clone: ~10% Lymphocytic/LPC clone: ~10% Hypocomplementemia 20% Negative tests for cryoglobulins | MGRS: 95% MM: 3%-4% WM/B-cell lymphoma: 1% |
| MGRS-related renal disorders without Ig deposits | |||||
| C3GP with monoclonal gammopathy | LM: MPGN, MesPGN, EPGN IF: granular deposits of C3 only in mesangium and CW EM: C3GN: less electron dense subendothelial/subepithelial/ mesangial deposits Dense deposit disease: highly electron dense sausage-like deposits within GBM | CKD (median s.creat.1.8 mg/dL) Proteinuria (median 3 g/d) Nephrotic syndrome (43%), Hematuria (90%) | Rare: Digital ischemia | Positive serum/urine EP/IFix: 100% Abnormal sFLC level: 50%-75% Plasma cell clone: 95% Lymphocytic clone: 5% Low C3 (35%-45%) High serum C5bC9: 80% | MGRS: 80%-90% MM: 5%-15% B-cell lymphoma: 6% |
| Thrombotic microangiopathy with monoclonal gammopathy | LM and EM: GBM duplication, mesangiolysis, subendothelial widening “fluff,” thrombosis IF: no Ig deposits | CKD (median s.creat 3 mg/dL) Proteinuria (median 2-4 g/d) Hematuria (90%) | Common: MAHA (50%-90%) CNS, skin, peripheral neuropathy | Positive serum/urine EP/IFix: 100%; Abnormal sFLC level: 28% Plasma cell clone: 80% Lymphocytic/LPC clone: 20% Low C3 (33%) High serum C5bC9: 77% | MGRS: 90% POEMS syndrome: 10% MM: 5% WM/B-cell lymphoma: 5% |
CH1, first constant domain; CNS, central nervous system; CW, glomerular capillary wall; EP, electrophoresis; EPGN, endocapillary proliferative glomerulonephritis; GBM, glomerular basement membrane; IF, immunofluorescence; IFix, immunofixation; LM, light microscopy; LPC, lymphoplasmacytic; MAHA, microangiopathic hemolytic anemia; MGN; membranous glomerulopathy; MesPGN, mesangial proliferative glomerulonephritis; MM, multiple myeloma; MPGN, membranoproliferative glomerulonephritis; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes; s.creat, serum creatinine; sFLC, serum free light chains; TBM, tubular basement membrane; WM, Waldenström macroglobulinemia.
Diagnosis of MGRS
The diagnosis of MGRS relies on the evaluation of clinical, pathological, immunological and hematological findings to establish the causal link between monoclonal gammopathy and kidney lesions.3
Clinical workup
MGRS should be suspected in any patient with MGUS, smoldering myeloma, or B-cell lymphoma presenting with renal symptoms, most commonly proteinuria. This underlines the crucial need for regular monitoring of proteinuria in the surveillance of MGUS. However, MGRS is frequently diagnosed in patients without known monoclonal gammopathy.
Determining the composition of proteinuria with electrophoresis analysis is the first step. Predominant albuminuria (urine albumin/total protein ratio >40%) indicates a glomerular disease, which has manifestations that vary among MGRS types. Massive albuminuria associated with full nephrotic syndrome and diffuse edema is frequent in AL amyloidosis, HCDD, ITGP, and PGNMID. Microscopic hematuria and hypertension are common, except in AL amyloidosis. Severe presentation, with acute kidney injury, hematuria, edema, and uncontrolled or malignant hypertension, is suggestive of cryoglobulinemic glomerulonephritis. Tests for cryoglobulins and serum complement levels provide diagnostic orientation. Except for AL amyloidosis and LCDD, all MGRS-related glomerulopathies may feature hypocomplementemia.3 Low serum C3 level only is a characteristic but inconstant finding in C3GP, TMA, and the LC variant of PGNMID.9-11,26
Tubular proteinuria with an albumin to protein ratio of less than 40% suggests LC proximal tubulopathy, with or without Fanconi syndrome. Almost invariably induced by the proximal tubule reabsorption of a monoclonal Vκ1LC, Fanconi syndrome manifests with slowly progressive CKD and symptoms of proximal tubulopathy (hypophosphatemia, hypouricemia, normoglycemic glucosuria, generalized aminoaciduria, low-molecular weight proteinuria, and proximal [type 2] tubular acidosis).18,19
Fanconi syndrome may be a manifestation of CSH, characterized by tissue infiltration with histiocytes featuring intracytoplasmic LC crystalline inclusions. In CSH, proximal tubule involvement often coexists with interstitial and perirenal fat infiltration.20
The second step is to carefully search for MIg-related extrarenal symptoms, with peculiar attention to cardiac, liver, and peripheral nerve involvement. Extrarenal disease is frequent in AL amyloidosis, cryoglobulinemias, and monoclonal immunoglobulin deposition disease (MIDD). Appropriate investigations are required, including measurements of serum cardiac biomarkers (troponin, Nt-proBNP/BNP), echocardiography, and/or cardiac magnetic resonance imaging.3 Diffuse bone pain secondary to hypophosphatemia-induced osteomalacia is common in Fanconi syndrome.18,19 CSH may manifest with widespread histiocytic infiltration of the lungs, gastrointestinal tract, cornea, bone marrow, lymph nodes, liver, and spleen.20 LC crystalline corneal deposition is frequent in LC crystalline podocytopathy, while skin lesions (rash, ulcers) are common in cryoglobulinemia and crystalglobulinemia.15-17
Pathological workup
A diagnostic kidney biopsy is required, except for AL amyloidosis already confirmed histologically (usually by lower lip, bone marrow, or other organ/tissue biopsy).3 The frequency of serious biopsy-related bleeding complications in MGRS appears similar to other indications (around 4%).27
Pathological studies include light microscopy on paraffin-embedded tissue (with systematic Congo red staining) and immunofluorescence on frozen tissue. Immunofluorescence is essential to assess the monotypic nature of deposits, ie, restriction to a single LC isotype and also a single heavy chain subclass if an entire MIg is involved. This requires a panel of reactive antibodies against IgG, IgM, IgA, kappa and lambda LC, and IgG subclasses if IgG deposits are present. EM is necessary for diagnostic confirmation, except in AL amyloidosis and LCDD.3
Paraffin immunofluorescence after protease digestion,28 immunofluorescence with heavy/light antibodies,29 immuno-EM, and mass spectrometry analysis after laser microdissection on paraffin-embedded tissue are sensitive techniques for the identification of MIg-related lesions.3,12,30 Available at specialized centers, these should be considered in difficult cases. Examples include false-negative frozen immunofluorescence results due to crystalline MIg deposits or apparent monotypic deposits despite negative testing for monoclonal gammopathy.3
CLINICAL CASE (continued)
Our patient displayed symptoms of glomerular disease in the setting of IgG kappa “MGUS” with abnormal kappa serum free light chain levels. Kidney biopsy revealed mesangial sclerosis with thickened tubular membranes. Immunofluorescence showed kappa LC–restricted linear deposits along glomerular capillary walls, tubular basement membranes, arterial myocytes, and in the mesangium, along with a powdery punctate appearance by EM, confirming the diagnosis of LCDD (Figure 3 a-f). A liver biopsy displayed similar linear kappa LC deposits in Disse's spaces.
Chasing the underlying clone
A complete hematologic workup is required to identify the monoclonal gammopathy, assess its concordance with kidney lesions (ie, matching with MIg deposits/inclusions), and characterize the underlying clonal disorder to deliver efficient therapy.
Monoclonal gammopathy testing relies on serum and urine protein electrophoresis and immunofixation, with serum FLC measurement. Because LCs are mainly degraded within the kidney, FLC levels rise as the GFR decreases (with an increased kappa to lambda ratio using the Freelite® assay) and should be interpreted accordingly. Although several FLC tests are currently available, their results are not interchangeable, and ideally the same test should be used in the same laboratory throughout follow-up.3
Detection and identification of the involved clone (ie, plasmocytic or lymphocytic) is based primarily on bone marrow studies with flow cytometry. Genetic studies (fluorescence in situ hybridization, NGS) are warranted to detect mutations of diagnostic (MYD88L265P if IgM monoclonal gammopathy), prognostic (t(4;14), del17p), or therapeutic interest (t(11;14)). IgG, IgA, and LC-only monoclonal gammopathies usually correspond to plasma cell clones and IgM to lymphocytic or lymphoplasmacytic clones.31 Exceptions exist, such as IgA or IgG-secreting lymphocytic clones of mucosa-associated lymphoid tissue (MALT) or chronic lymphocytic leukemia (CLL) type or t(11;14)-positive IgM-secreting plasma cell clones.3,32
In patients without a detectable bone marrow clone, flow cytometry of peripheral blood lymphocytes and imaging studies (computed tomographic and/or positron emission tomographic scan, whole-body magnetic resonance imaging) should be considered to identify a small CLL-like population, low-grade lymphoma, or localized plasmacytoma.3,31
Overall, a monoclonal gammopathy and corresponding clonal disorder is detected in 80% to 100% of MGRS renal disorders. Around 10% of patients with AL amyloidosis and renal-limited or predominant disease have normal FLC levels at diagnosis. The pathogenic clone is composed of plasma cells in most MGRS types, whereas lymphocytic proliferations are frequent in ITGP (up to 80% of cases) and type 1 cryoglobulinemic glomerulonephritis (50%).13-15
The clone detection rate in PGNMID differs from other MGRS- related nephropathies: only 30% of patients have abnormal immunofixation and/or FLC tests, and less than 25% have a detectable plasma cell or lymphocytic disorder by bone marrow/blood flow cytometry analysis.24-26 Rates are particularly low (around 10%) in PGNMID with IgG3 deposits. The occurrence of PGNMID-IgG3 in young patients or after an infectious episode raises suspicion that some cases may not correspond to MGRS but may derive from oligoclonal IgG deposition secondary to a skewed B-cell repertoire in response to viral or other antigenic triggers.26
Recently, RNA-based Ig repertoire sequencing has emerged as a highly sensitive tool for detecting subtle B-cell clones in MGRS patients with low or normal serum MIg levels and negative bone marrow studies.33 The development of such molecular approaches, coupled with mass spectrometry analysis of the circulating and deposited MIg, may facilitate the diagnosis and improve the management of MGRS in the near future.
CLINICAL CASE (continued)
In the present patient, the presence of a small IgG kappa–secreting plasma cell clone was consistent with a final diagnosis of MGRS-related kappa LCDD with renal and liver involvement.
Management of MGRS
Outside of AL amyloidosis, evidence-based recommendations for treating MGRS are limited. Current treatment is based on suppressing the pathogenic MIg secretion through clone- targeted therapy. The use of agents with renal elimination (melphalan and lenalidomide) or potential nephrotoxicity (eg, carfilzomib-associated TMA) should be avoided.3,31 For plasma cell clones, current regimens are based on bortezomib and/or anti-CD38 monoclonal antibodies. These combinations provide high rates of deep hematologic response with a good tolerance profile, even in patients with severe CKD or end-stage kidney disease (ESKD) requiring chronic dialysis.3,34,35 Consequently, the indications for high-dose therapy and stem cell transplantation have decreased. Venetoclax does not require dose adaptation to kidney function and appears a promising option in MGRS involving t(11;14) plasma cells.36 The risk for tumor lysis syndrome limits its use in patients with CCL-like clones. Treatment of MGRS with lymphocytic/lymphoplasmacytic clones relies primarily on rituximab-based combinations. When a clone is not identified, the choice of regimen is empiric, based on the characteristics of monoclonal gammopathy (see above).3,31 Complementary approaches with immunotherapy aimed at favoring the elimination of tissue fibrils are under investigation in AL amyloidosis.
The decision to treat requires a careful evaluation of the benefit-to-risk balance, considering the severity of kidney disease, extrarenal manifestations, and comorbidities. The introduction of a triplet or quadruplet regimen is logical if a renal response is possible (ie, eGFR >15 mL/min/1.73 m2) and/or if other organ/tissue involvement is present (eg, amyloid cardiopathy). In patients with ESKD, indications are extrarenal involvement or the prevention of disease recurrence in the allograft in those eligible for renal transplantation.3,31,37,38
The baseline eGFR level and the deepness of the hematologic response are the main determinants of long-term outcomes.3,10,13-15,21-23,39,40 The regular assessment of clonal response after each cycle to adapt the therapeutic regimen and throughout follow-up to detect early hematologic relapse or progression is mandatory. International criteria for hematologic response in AL amyloidosis, based on FLC measurements, (Table 3) are commonly used for other LC-related MGRS.34,39 In nephropathies involving an entire MIg, the evaluation of hematologic response based on electrophoresis (monoclonal spike) and immunofixation is often difficult.3,31
Definitions for hematologic response in AL amyloidosis
| Response categories | Definitions |
|---|---|
| Complete responsea | Negative serum and urine immunofixation electrophoresis and normalization of FLC ratio (or uninvolved FLC concentration greater than involved FLC concentration with or without an abnormal FLC ratio) |
| Very good partial response | If baseline dFLC ≥50 mg/L: reduction in dFLC <40 mg/L |
| Partial response | If baseline dFLC ≥50 mg/L: reduction in dFLC >50% If baseline dFLC 20-50 mg/L: reduction in dFLC <10 mg/L |
| No response | All other patients |
| Response categories | Definitions |
|---|---|
| Complete responsea | Negative serum and urine immunofixation electrophoresis and normalization of FLC ratio (or uninvolved FLC concentration greater than involved FLC concentration with or without an abnormal FLC ratio) |
| Very good partial response | If baseline dFLC ≥50 mg/L: reduction in dFLC <40 mg/L |
| Partial response | If baseline dFLC ≥50 mg/L: reduction in dFLC >50% If baseline dFLC 20-50 mg/L: reduction in dFLC <10 mg/L |
| No response | All other patients |
A deep hematologic response—that is, a very good partial response (VGPR) defined by a difference between involved and noninvolved FLC (dFLC) of less than 40 mg/L—or a complete response is an independent predictor of renal and patient survival in MGRS.10,21-23,39 Clone-targeted therapy should not be overly prolonged after achieving a response, as renal response is usually delayed. The latter is based on serum creatinine, eGFR, 24-hour proteinuria, or the urine protein to creatine ratio, which should be monitored at each treatment cycle and on regular intervals thereafter (at least every 3 months).3,31 Other factors determine renal outcomes, including the extent of interstitial fibrosis/tubular atrophy on kidney biopsy and eGFR at diagnosis. A baseline eGFR value greater than or equal to 50 mL/min/1.73 m2 (with proteinuria <5 g/24 h) in AL amyloidosis and greater than or equal to 30 mL/min/1.73 m2 in LCDD is associated with improved renal survival.21-23,40
Renal supportive care
Controlling hypertension and fluid retention relies on salt restriction and diuretics. Renin angiotensin system blockers are commonly used to reduce albuminuria, but their tolerance is poor in AL amyloidosis–associated cardiopathy or autonomic dysfunction. The benefit of SGLT2 inhibitors in patients with proteinuria and/or heart failure appears promising but requires confirmation. Prophylactic anticoagulation is indicated in glomerular disorders featuring severe hypoalbuminemia, considering the increased hemorrhagic risk in AL amyloidosis.3,31
Both hemodialysis and peritoneal dialysis can be proposed to patients with ESKD. Peritoneal dialysis may be better tolerated in those with severe heart disease or dysautonomia, without an increased infectious risk compared to hemodialysis.3 In eligible patients without severe extrarenal disease, renal transplantation is the treatment of choice. A VGPR or CR before the procedure is associated with favorable allograft and patient survival, as shown in AL amyloidosis and MIDD.21,37,38 Recurrence remains a significant cause of allograft loss and morbidity. The risk, variable across MGRS types, appears higher in C3GN and PGNMID.38 Careful prolonged hematologic surveillance is required. Recurrence should be confirmed histologically and treated to preserve allograft function.
CLINICAL CASE (continued)
The patient received frontline therapy with daratumumab-bortezomib-dexamethasone. After 6 cycles, he achieved a VGPR (dFLC, 1.3 mg/dL; monoclonal spike 0.3 g/dL), with liver and renal response (proteinuria, 62 mg/mmol; eGFR, 45 mL/min/1.73 m2).
Conflict-of-interest disclosure
Frank Bridoux: honoraria: Amgen, Janssen, GSK, Lilly, Sanofi; advisory board: Novartis, AstraZeneca, Attralus.
Samih H. Nasr: no competing financial interests to declare.
Bertrand Arnulf: honoraria: Janssen, Bristol Myers Squibb, Sanofi, GSK; research funding: Bristol Myers Squibb; advisory board: Janssen, Bristol Myers Squibb, Takeda.
Nelson Leung: research funding: Omeros; stockholder: AbbVie, Checkpoint Therapeutics.
Christophe Sirac: research funding: Attralus, Gate Bioscience, Amyl Therapeutics.
Arnaud Jaccard: honoraria: Janssen, Celgene, Amgen, Pfizer; research funding: Celgene, Janssen, Sanofi; advisory board: Janssen, Prothena.
Off-label drug use
Frank Bridoux: nothing to disclose.
Samih H. Nasr: nothing to disclose.
Bertrand Arnulf: nothing to disclose.
Nelson Leung: nothing to disclose.
Christophe Sirac: nothing to disclose.
Arnaud Jaccard: nothing to disclose.
